Publié le 15 jan 2013Lecture 7 min
Rétinoblastome de l'enfant : comment le repérer ?
L. DESJARDINS, Institut Curie, Paris
Le rétinoblastome est une tumeur rare dont l’incidence est de 1/15 000-20 000 naissances par an, mais c’est la tumeur maligne intraoculaire la plus fréquente chez l’enfant. Dans environ 60 % des cas, le rétinoblastome est unilatéral avec un âge médian au diagnostic de 2 ans. Dans 40 % des cas, il est bilatéral avec un âge médian au diagnostic de 1 an. Toutes les formes bilatérales et unilatérales multifocales sont héréditaires. La fréquence des formes familiales est actuellement en augmentation.
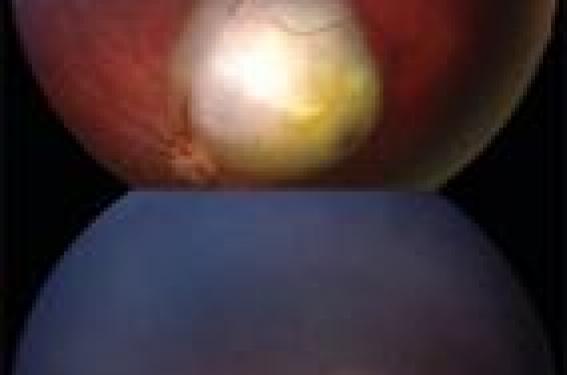
Bases génétiques Le gène du rétinoblastome (Rb1) a été le premier gène suppresseur de tumeur identifié. En 1971, Knudson a émis l’hypothèse selon laquelle deux événements génétiques sont nécessaires au développement du rétinoblastome. Une perte ou une mutation des deux allèles du gène Rb1, localisé au niveau du chromosome 13q1.4 est nécessaire pour que la tumeur survienne. Dans les formes héréditaires, représentant environ 50 % des cas, les patients sont porteurs d’une mutation germinale, présente dans toutes les cellules de l’organisme, et d’une altération somatique du gène Rb1 présente uniquement au niveau des cellules rétiniennes tumorales. Les mutations germinales de Rb1 concernent tous les patients atteints de forme bilatérale et 15 à 20 % des formes unilatérales. La mutation se transmet selon un mode autosomal dominant bien que le gène fonctionne selon un mode récessif. La pénétrance est d’environ 90 %. Dans les formes sporadiques, représentant environ 50 % des patients, il existe une inactivation somatique des deux allèles du gène Rb1. Dans ce cas, le rétinoblastome est toujours unilatéral et unifocal. Le rétinoblastome se développe plutôt vers le centre de la rétine au cours des premiers mois de vie et plutôt de plus en plus vers la périphérie de la rétine au fur et à mesure que l’enfant grandit. Après 8 ou 9 mois, les nouvelles tumeurs sont en général très périphériques. De ce fait dans les formes familiales pour lesquelles un dépistage mensuel par examen systématique du fond d’oeil est réalisé, il faut faire un examen soigneux de la périphérie rétinienne en indentant la périphérie et en vérifiant l’ora serrata (extrême périphérie rétinienne) sous anesthésie générale. Figure 1. Tumeur du pôle postérieur détectée précocement à 6 mois en raison d’un strabisme (A). Même tumeur après traitement conservateur (B). Ce dépistage est réalisé de façon systématique chez tous les enfants à risque, c'est-à-dire ceux dont les parents ont présenté un rétinoblastome bilatéral ou unilatéral et également ceux qui ont eu un frère ou une soeur, ou un autre membre de la famille atteint. La consultation de génétique permet de proposer une étude constitutionnelle du gène Rb chez la personne atteinte et, si la mutation est identifiée, de savoir si l’enfant est ou non porteur de cette mutation et d’adapter ainsi le suivi : pas de suivi pour les enfants non porteurs ; suivi mensuel et dès les premiers jours de vie pour les enfants porteurs. En l’absence de mutation identifiée, la fréquence du suivi sera adaptée à l’importance du risque. Une étude génétique indirecte permet parfois de dire que deux frères ou soeurs ne sont pas porteurs du même allèle et de lever la surveillance. Diagnostic clinique Les signes révélateurs les plus fréquents sont le strabisme et la leucocorie. Le strabisme est un signe parfois précoce. En effet, la survenue d’une tumeur dans le centre de la rétine peut altérer la vision et provoquer un strabisme unilatéral. Comme nous l’avons dit précédemment, c’est au cours des premiers mois de vie que peut survenir une tumeur maculaire. À cet âge, il existe fréquemment un strabisme accommodatif qu’il faut bien différencier du strabisme symptôme d’une affection organique de la rétine. Le strabisme accommodatif est un strabisme convergent, bilatéral et intermittent, alors que le strabisme lié à une affection organique est unilatéral et permanent. Quelquefois, cependant, le strabisme est plus marqué à la fatigue. De toute façon, il est très important de consulter un ophtalmologiste quel que soit le type de strabisme. L’ophtalmologiste pourra même chez un nourrisson vérifier la vision et surtout pratiquer un examen du fond d’oeil s’il constate effectivement un strabisme ou une mauvaise vision d’un oeil. La règle pour les ophtalmologistes est l’examen du fond d’oeil chez tout enfant strabique. Il faut distinguer un strabisme accomodatif d’un strabisme symptômatique d’une affection organique. Grâce à cet examen précoce du fond d’oeil, on peut parfois diagnostiquer des tumeurs de petite taille qui sont accessibles à un traitement conservateur de l’oeil (figure 1). La leucocorie À un stade plus tardif, le signe essentiel du rétinoblastome est la leucocorie (figure 2), qui peut être isolée ou associée à une déviation strabique. La leucocorie est un reflet blanc dans la pupille encore appelé « oeil de chat amaurotique ». Figure 2. Strabisme et leucocorie. Elle est parfois bien visible sur des photos au flash (si on n’utilise pas le système anti-yeux rouges, on a une pupille orangée quand le fond d’oeil est normal et blanche en cas de leucocorie). La leucocorie peut être au début inconstante, visible seulement sous certains éclairages et dans certaines directions du regard, notamment pour les tumeurs périphériques. C’est un signe important qu’il ne faut pas négliger sous peine de retard au diagnostic. Il faut savoir écouter les parents lorsqu’ils décrivent ce symptôme même si on ne retrouve pas la leucocorie au cabinet. Il faut néanmoins savoir qu’au stade de leucocorie, le rétinoblastome est souvent très volumineux occupant une bonne partie de la cavité vitréenne (intérieur de l’oeil). Le diagnostic est alors important pour éviter un envahissement du nerf optique, de la chambre antérieure ou extrascléral, qui sont des facteurs de gravité de la maladie ; mais un traitement conservateur de l’oeil à ce stade n’est souvent plus possible. Autres signes Si la maladie n’est pas diagnostiquée au stade de strabisme ou de leucocorie, d’autres symptômes peuvent apparaître. Ils témoignent en général de formes plus évoluées et plus graves qui nécessitent des traitements plus lourds. • La buphtalmie est une augmentation du volume du globe oculaire liée à une distension de la sclère sous l’effet de la pression exercée par la tumeur. Le globe oculaire apparaît plus volumineux avec une augmentation du diamètre cornéen et parfois un aspect bleuté de la sclère amincie. • Dans d’autres cas, la tumeur peut s’accompagner de réactions inflammatoires au niveau de l’oeil et parfois même de l’orbite avec gonflement, rougeur et douleurs. • Un changement de la couleur de l’oeil est parfois noté • Enfin, en l’absence de diagnostic, on peut avoir à faire à des formes évoluées qui actuellement sont beaucoup plus fréquentes dans les pays en voie de développement. La tumeur envahit dans ce cas les tissus orbitaires avoisinants provoquant une exophtalmie dans un contexte inflammatoire et douloureux. Conclusion Le diagnostic précoce du rétinoblastome repose sur la meilleure connaissance des signes évocateurs du rétinoblastome que sont le strabisme et la leucocorie. La prise en compte rapide de ces anomalies permet de préserver l’oeil et la vision et bien sûr d’éviter de mettre en jeu le pronostic vital.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :