Publié le 18 jan 2021Lecture 6 min
Un ictère néonatal prolongé
Marc SZNAJDER, Paris

César est né le 24 septembre 2019 de parents non consanguins, au terme d’une grossesse primipare primigeste, à 40 semaines d’aménorrhée. Il n’y a pas d’antécédent familial particulier, pas de maladie hépatobiliaire, pas de lithiase biliaire, ni de cholestase gravidique. PN : 3 790 g ; TN : 51 cm ; PCN : 34 cm ; APGAR : 10-10. Il n’est pas noté de signe de souffrance fœtale aiguë, ni de bosse sérosanguine, ni de problème de groupe sanguin. César est alimenté d’emblée au lait artificiel.
Un ictère modéré apparaît progressivement dans les premiers jours. L’enfant est simplement surveillé en maternité, il n’inspire pas d’inquiétude.
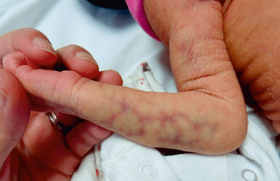
Au 10e jour de vie, cet ictère cutanéo-muqueux apparaît modéré cliniquement. César grossit bien, il pèse 3 750 g, ses selles et ses urines sont jugées normales, de même que le reste de son examen. On ne constate ni hépatosplénomégalie, ni dysmorphie.
À un mois de vie, l’ictère est identique, les selles ne sont pas jugées décolorées, l’enfant grossit normalement (P = 4 280 g ; T = 55 cm ; PC = 37 cm).
Un 1er bilan est alors pratiqué en ville :
– échographie abdominale (H2 après biberon) : foie normal, voies biliaires intrahépatiques non dilatées, cholédoque normal, vésicule bien vue, pas de lithiase ;
– biologie : Hb : 14,2 g/dl ; VGM : 100 µm3 ; GB et plaquettes normaux ; CRP < 1 mg/l ;
– bilirubine totale : 181,1 mol/l (105,9 mg/l), bilirubine conjuguée 137,9 mol/l (80,6 mg/l) ; T4 (16,5 pmol/l) et TSH (0,984 mUI/l) normales ; ECBU normal.
L’existence d’un ictère cholestatique est donc confirmée. Un nouveau bilan pratiqué à 6 semaines note un poids à 4 650 g et un ictère stable chez un enfant en bon état général. Le bilan biologique s’est altéré :
– cytolyse hépatique : ASAT : 506 U/l (> 10 N), ALAT : 222 U/l (5N) ;
– cholestase : PAL : 633 UI/l, GGT : 126 UI/l ;
– ictère à bilirubine mixte à prédominance conjuguée (bilirubine totale : 106, conjuguée 83 mol/l) ;
– pas d’insuffisance hépatique (TP : 91 %); albumine Nl (43,4 g/l) ;
– CPK : 110 U ;
– alpha 1 anti-trypsine (AAT) : 1,29 g/l (N) ;
– acides biliaires : 57,3 (n < 5/l) ; Hb : 10,4 g/dl, VGM : 95 µm3.
L’enfant est initialement hospitalisé : les selles sont alors jugées partiellement décolorées, plutôt jaune pâle depuis le début (ce que la maman conteste !) ; on retrouve une hépatomégalie de + 3 cm en sous-costal droit, non ferme, sans splénomégalie, ni ascite ; le faciès est normal. On constate cette fois-ci de petite vésicule biliaire à l’échographie, sans autre anomalie.
Devant cet ictère cholestatique à gamma GT élevées, le diagnostic d’atrésie des voies biliaires extrahépatiques (AVBEH) est d’emblée suspecté, et les principaux diagnostics différentiels sont écartés :
– mucoviscidose (test sueur et trypsine immuno-réactive normaux) ;
– déficit en alpha antitrypsine : 1,5 g/l (N) ;
– syndrome d’Alagille : pas de dysmorphie faciale, pas de vertèbres en ailes de papillon.
L’échographie cardiaque est normale (pas de sténose pulmonaire), pas d’embryotoxon ;
– déficit de synthèse des acides biliaires : non car les taux sont augmentés ;
– maladie métabolique : glycémie normale, ammoniémie 67 umol/l : rapport lactate/pyruvate normal, (CAA et CAO en cours) ;
– cause infectieuse : ECBU normal, PCR VHB, VHC, CMV, EBV, adénovirus, HHV6, parvovirus négatifs ; CMV urinaire négatif ;
– enquête génétique non faite à ce stade.
César reçoit un hydrolysat de PLV (Prégestimil®) enrichi en dextrine maltose, de l’Ursofalk® (acide ursodésoxycholique, acide biliaire naturel), une supplémentation vitaminique (ADEK), et est transféré en service d’hépatologie infantile pour suspicion d’AVBEH à 7 semaines de vie.
À l’arrivée dans ce service :
– l’ictère est jugé stable, chez un enfant qui grossit moins bien (P = 4 800 g) ;
– l’échographie hépatique note une vésicule biliaire petite, à paroi irrégulière, pas correctement distendue après 4,5 h de jeûne, compatible avec le diagnostic d’AVBEH. Toutefois, on ne met pas en évidence de « triangular cord sign » (résidu fibreux du canal biliaire extrahépatique, signe très spécifique d’atrésie des VB) ;
– le bilan ophtalmologique complet n’apporte pas d’argument en faveur d’un syndrome d’Alagille (pas d’embryotoxon).
Devant ce doute diagnostique, César est transféré en chirurgie pour laparatomie exploratrice, cholangiographie et intervention de Kasaï (à M2) le cas échéant :
– la cholangiographie infirme finalement le diagnostic d’AVBEH ;
– la biopsie hépatique permet le diagnostic de ductopénie majeure (> 80 %), avec cholestase histologique intense, nombreux hépatocytes géants plurinucléés, fibrose à contours étoilés.
En post-opératoire, on observe une régression progressive de l’ictère, qui va disparaître complètement et une amélioration du bilan hépatique. Une introduction partielle du lait 1er âge (50 %) est réalisée.
À l’âge de 4 mois, l’échographie abdominale montre un foie de taille normale, aux contours lisses, homogènes et l’élastométrie* hépatique est normale (médiane : 7,2 kP).
Le bilan étiologique de cette ductopénie non syndromique est complété par la recherche par NGS (séquençage de nouvelle génération) de cholestases génétiques à GGT élevées (dont 2 mutations associées au syndrome d’Alagille, JAG1 et NOTCH2), ainsi qu’une chromatographie des acides gras à très longue chaîne, un dosage de l’oxystérol et des lysosphingolipides (résultats en attente).
Le traitement par Ursofalk® est poursuivi : 600 mg/m2/j + supplémentation vitaminique injectable tant que la bilirubine totale > 50 µmol/l, mais rapidement celle-ci revient à 15 µmol/l et le relais est pris par ADECK per os. César se développe normalement, sans ictère clinique.
Discussion
La cholestase néonatale survient chez environ 1 sur 2 500 nouveaunés à terme : les causes les plus fréquentes étant l’AVB (50 % des cas), les infections virales et le déficit en α1 antitrypsine.
L’atrésie des voies biliaires (AVB)
L‘AVB est le premier diagnostic à évoquer devant une cholestase néonatale prolongée audelà de 10 jours de vie. En effet, le délai diagnostique et la date d’intervention chirurgicale y sont des facteurs pronostiques majeurs, pour prévenir autant que possible le risque de cirrhose biliaire : bien que rare, l’AVB est l’indication principale de transplantation hépatique chez l’enfant.
Classiquement, elle se démasque rapidement par des selles décolorées. Cependant, cette décoloration peut être partielle, la surface des selles peut être teintée par des urines ictériques. Le risque de confusion existe aussi avec les hydrolysats de protéines lactées.
Une échographie abdominale doit être effectuée rapidement chez tous les nourrissons cholestatiques par un(e) radiologue expérimenté(e). Lorsque les canaux biliaires intrahépatiques sont dilatés, on suspecte surtout des calculs biliaires ou un kyste du cholédoque. Une vésicule biliaire petite et dysmorphique évoque très fortement une AVB mais n’est pas spécifique, car la vésicule peut être vide quand la cholestase est sévère. Au contraire, une vésicule normale n’exclut pas une AVB car il existe des formes kystiques de cette anomalie.
Une publication récente fait état de l’intérêt de l’élastométrie hépatique pour discriminer l’AVBEH (en faveur de ce diagnostic si > 7,7 kPa) des autres causes de cholestases néonatales(1). Une autre étude radiologique pointe la valeur du « triangular cord sign », de l’élargissement de l’artère hépatique, des signes d’hypertension portale et de la non-visualisation de la voie biliaire principale, comme signes en faveur d’une AVB par rapport à une ductopénie, alors qu’une petite vésicule biliaire se voit dans les deux cas(2).
Pour sensibiliser parents et praticiens à ce sujet, une échelle colorimétrique des selles est désormais disponible dans les carnets de santé.
Dans notre observation, le diagnostic d’AVB, jugé probable initialement, a finalement été écarté au profit de celui de ductopénie, non syndromique puisqu’aucune des manifestations extrahépatiques du syndrome d’Alagille (embryotoxon, vertèbres en ailes de papillon, sténose pulmonaire, faciès évocateur) n’a été retrouvée.
La paucité ductulaire ou ductopénie, syndromique ou non syndromique, est une cause rare de cholestase prolongée. Elle est due à l’absence partielle ou totale des canaux biliaires intrahépatiques, en relation avec une anomalie génétique (cf. plus haut).
Le diagnostic est histologique, il montre une disparition des canaux biliaires intrahépatiques interlobulaires et septaux dans au moins 50 % des espaces portes, sur une biopsie hépatique qui doit en contenir au moins 10. À noter que dans l’AVB, on observe au contraire une prolifération ductulaire réactionnelle.
Le bilan étiologique comprend une enquête médicamenteuse, un bilan viral, métabolique, autoimmun, génétique et une imagerie des voies biliaires intra- et extrahépatiques. La ductopénie est dite « idiopathique » lorsque ce bilan est négatif.
Le pronostic de la ductopénie peut être péjoratif, lié au risque d’évolution vers la cirrhose biliaire (30 % des cas ?).
Figure 2. Cholestase néonatale : conduite pratique et étapes du diagnostic(5)
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :