Publié le 18 jan 2021Lecture 9 min
Conduite à tenir devant une sclérodermie juvénile
Anne WELFRINGER-MORIN, Service de dermatologie du Pr Bodemer, centre de référence des Maladies de la peau et des muqueuses d’origine génétique (MAGEC), hôpital Necker-Enfants malades, Paris

La sclérodermie juvénile est une maladie rare du tissu conjonctif caractérisée par une inflammation, des anomalies vasculaires et une fibrose pouvant toucher la peau, mais aussi les organes internes dans le cadre d’une atteinte systémique. Elle peut être localisée ou systémique, le passage de la forme localisée à systémique étant exceptionnelle. La forme la plus fréquente est la sclérodermie juvénile localisée. Elle touche principalement les filles.
Sclérodermie juvénile localisée
Elle est définie par un épaississement et une induration localisée à la peau, mais elle peut toucher les tissus plus profonds allant de la graisse sous-cutanée, aux muscles, au périoste et à l’os. Elle s’observe principalement chez les filles avec un âge moyen de début de 7,3 ans(1). Il existe différents types de sclérodermies localisées.
La sclérodermie en plaque ou morphée
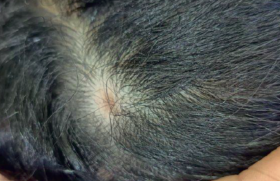
Elle touche principalement le tronc. Cliniquement, elle débute par une lésion généralement maculeuse circonscrite, érythémateuse ou brune lilacée, souple (figure 1) avec une extension centrifuge. Cette lésion devient secondairement scléreuse, blanche, nacrée au centre avec un possible halo érythémateux (« lilac ring ») lorsqu’elle reste évolutive. Au stade séquellaire (figure 2), la lésion est lisse, dépilée, atrophique et anhidrotique, hypo- ou hyperpigmentée, soupe au palpé ou encore scléreuse.
Figure 1. Sclérodermie en plaque ou morphée.
Figure 2. Lésion de sclérodermie en plaque au stade séquellaire.
La sclérodermie en bandes ou linéaire (figure 3)
C’est le sous-type le plus fréquent. La gravité de cette sclérodermie est son extension possible en profondeur. Elle est souvent active plusieurs années avec extension. Les localisations préférentielles sont les membres (83 %) puis la tête (17 %). À la phase aiguë, ces sclérodermies peuvent être associées à des manifestations musculaires (myosite) ou articulaires (raideur articulaire, arthrite). Une forme particulière est à connaître : la sclérodermie en coup de sabre (figure 4) touchant plus particulièrement la région paramédiane du front, du cuir chevelu au sourcil avec une alopécie cicatricielle. Les complications possibles sont des troubles visuels ou neurologiques associés, des hypoplasies osseuses, des malpositions dentaires, des séquelles esthétiques. L’hemi-atrophie de Parry Romberg est définie par une atrophie initiale majeure et évolutive, atteignant inexorablement une hémiface avec atrophie musculaire, osseuse et du tissu graisseux.
Figure 3. Sclérodermie en bandes ou linéaire.
Figure 4. Sclérodermie en coup de sabre.
Les morphées généralisées
Cette forme est définie par la présence d’au moins 4 lésions ou plus, de grande taille (> 3 cm) touchant plus de 2 sites anatomiques différents. Elle est de mauvais pronostic et résistante au traitement. Les atteintes extra-cutanées des sclérodermies localisées sont plus fréquentes chez l’enfant (20 %), notamment dans les formes linéaires(2). L’atteinte la plus fréquente est musculosquelettique avec, en cas d’atteinte monomélique de membre, un possible engainement articulaire locorégional source d’un retentissement fonctionnel, et d’un raccourcissement parfois sévère du membre avec amyotrophie sévère. Le bilan minimal à réaliser devant une morphée comprend :
une numération formule sanguine montrant souvent une hyperéosinophilie en phase active ;
un bilan inflammatoire avec, dans 30 % des cas, une augmentation de la VS et de la CRP en phase active ;
un bilan auto-immun révélant un FR (facteur rhumatoïde) positif dans 25 à 40 % des cas, des AAN (anticorps antinucléaires) 30 à 76 % des cas et une absence d’anticorps anti-Scl70 et anticentromères.
Le bilan morphologique minimal comporte un examen ophtalmologique systématique et une exploration neurologique (imagerie cérébrale et électro-encéphalogramme) en cas de morphée linéaire du visage. Les autres examens dépendent de la clinique, notamment le recours à l’IRM des parties molles en cas de suspicion d’extension en profondeur.
Traitement
L’approche thérapeutique n’est pas codifiée et dépend de l’âge du patient, du retentissement fonctionnel (atteinte des tissus profonds ou des articulations) et esthétique, de l’activité de la maladie et de la zone lésionnelle(3). Il n’existe pas de traitement spécifique pour lequel une efficacité est assurée. La majorité des traitements proposés sont issus de publications concernant des cas ponctuels et/ou petites séries avec de rares études prospectives en double aveugle contre placebo(4). Les données sont difficiles d’interprétation en l’absence d’un moyen d’évaluation objectif reproductible et validé de l’activité de la maladie. Les morphées peuvent régresser spontanément, inconstamment. Les formes linéaires se stabilisent spontanément après quelques années, mais avec un risque de séquelles quand elles ont évolué en profondeur. Les hémi-atrophies de Parry Romberg sont à ce jour au-delà des ressources thérapeutiques curatives et les lésions séquellaires pourront être prises en charge en chirurgie, notamment par injections de graisses autologues. Les stratégies thérapeutiques reposent sur un traitement local en cas de morphée circonscrite, principalement les dermocorticoïdes de classe IV.
En cas de morphée linéaire avec risque d’extension en profondeur et de séquelles, le traitement le plus fréquemment utilisé, repose sur un traitement de fond par méthotrexate associé à une corticothérapie per os sur une moyenne de 3 à 6 mois. En cas d’échec, une discussion avec une équipe experte est indispensable pour chercher des alternatives : un traitement par mycophénolate mofétil pourra être proposé. Chaque stratégie thérapeutique évalue soigneusement le rapport bénéfice/risque. La prise en charge par un kinésithérapeute, à visée d’assouplissement de la peau, de lutte contre la raideur et les rétractions articulaires, est la pierre angulaire du traitement. La durée moyenne de la maladie est longue et les rechutes sont fréquentes parfois à distance(5). Les complications sont cutanées (atrophie, ulcérations, poikilodermie, etc.), musculaires (atrophie, myosite rétractile), articulaires et osseuses. En effet, les enfants avec une sclérodermie juvénile localisée sont à risque de perturbations de la croissance (différences de longueur des membres) et d’atrophie faciale. L’accompagnement psychologique est indispensable dans ces formes sévères avec retentissement fonctionnel et esthétique.
Sclérodermie systémique
Seulement 3 % des sclérodermies systémiques ont un début de la maladie dans l’enfance. C’est la troisième pathologie rhumatismale de l’enfant, après l’arthrite juvénile idiopathique et le lupus érythémateux systémique. Elle touche principalement les filles à un âge moyen de 8,1 ans. Le début de la maladie est in - sidieux expliquant un retard diagnostique d’environ 2 ans. On classe la sclérodermie systémique en 3 groupes (selon Leroy) :
sclérodermie systémique cutanée diffuse (la sclérose cutanée remonte au-dessus des coudes et/ou des genoux) ;
sclérodermie systémique cutanée limitée (la sclérose cutanée ne remonte pas au-dessus des coudes et des genoux) ;
sclérodermie systémique sine scleroderma (en l’absence de sclérose cutanée).
Dans les nouveaux critères de classification ACR-EULAR, la présence d’un épaississement cutané des doigts des mains audelà des articulations métacarpo-phalangiennes permet de classer le patient comme atteint de sclérodermie systémique. En l’absence de ce critère, 7 critères alternatifs aident au diagnostic : des lésions pulpaires, des télangiectasies, des anomalies à la capillaroscopie, une atteinte pulmonaire, un phénomène de Raynaud et la présence d’anticorps spécifiques de la sclérodermie. Cette classification est adaptée à l’adulte et ne peut s’appliquer stricto sensu chez l’enfant. Le phénomène de Raynaud est le premier signe de la maladie, il peut précéder les autres symptômes de 19 mois. Il a comme particularité d’être asymétrique et pouvant survenir l’été. Il s’accompagne de modifications pulpaires avec hyperkératose. Il peut se compliquer d’ulcérations digitales (présent dans 10 % des cas au diagnostic) (figure 5), cicatrisant difficilement avec risque de surinfection. Le bilan minimal devant un phénomène de Raynaud chez l’enfant nécessite une recherche de FAN (facteur antinucléaire) et une capillaroscopie, avec une fréquence de suivi à adapter en fonction des résultats (6).
Figure 5. Ulcérations digitales en rapport avec un phénomène de Raynaud.
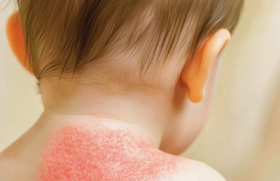
L’atteinte cutanée se manifeste par un premier stade œdémateux (figure 6) (ferme, indolore, ne prenant pas le godet) qui évolue secondairement par un stade fibreux avec une induration de la peau puis vers un stade atrophique. La sclérose s’étend de façon progressive et symétrique du segment distal (sclérodactylie, acrosclérose) vers le proximal. La fibrose peut progresser en profondeur entraînant des rétractions articulaires. Ceci peut s’accompagner de poikilodermie et de télangiectasies. La fibrose donne un aspect figé et inexpressif du visage. Les calcinoses touchent 1/5 des patients (figure 7).
Figure 6. Stade œdémateux de la sclérodermie systémique.
Figure 7. Calcinose chez un patient avec sclérodermie systémique.
L’atteinte articulaire est la plus fréquente des atteintes extracutanées chez l’enfant et touche un tiers des patients. Les manifestations musculo-articulaires peuvent être précoces et précéder les lésions cutanées. Des connectivites de chevauchement sont souvent observées. L’atteinte digestive peut toucher l’ensemble du tube digestif, de la cavité buccale à la terminaison anorectale. La plus fréquente est l’atteinte œsophagienne qui touche trois quarts des enfants avec un reflux gastro-œsophagien et des troubles de la motilité. Les lésions respiratoires s’observent chez 20 % des patients et sont marquées par une diminution de la capacité vitale ou une diminution de la DLCO aux EFR, avec une possible atteinte fibrosante au TDM thoracique. L’atteinte cardiaque est rare, mais sévère, et est la principale cause de mortalité chez l’enfant(7). Contrairement à l’adulte, la crise rénale sclérodermiforme, est exceptionnelle.
Le bilan biologique standard et auto-immune recherche une atteinte d’organe. Il vise à mettre en évidence une inflammation sanguine, un syndrome de ma - labsorption, des signes de myosite et une atteinte rénale. Sur le plan auto immun(8), les particularités chez l’enfant sont la faible présence d’anticorps anti-centromères, la rareté des anticorps antiARN polymérase III (associés à l’atteinte rénale) et la fréquence importante d’anticorps anti-PMScl, anti-UIRNP que l’on retrouve dans les syndromes de chevauchement. Le bilan systémique minimal comprend des EFR avec une imagerie pulmonaire, une évaluation cardiaque (ECG, échographie cardiaque ± IRM cardiaque), une exploration digestive en fonction des symptômes (± Phmétrie ou manométrie systématique). Un score de sévérité a été proposé pour guider la prise en charge : le « Juvenile Systemic Sclerosis Severity Score » incluant l’atteinte cutanée, les différentes atteintes d’organes et la croissance(9).
Traitement
Il repose sur l’atteinte d’organe. En cas d’atteinte uniquement cutanée et musculo-articulaire, le méthotrexate est le traitement de première intention avec des corticoïdes en cas de poussée. Le traitement de l’atteinte vasculaire repose sur les inhibiteurs calciques et la protection du froid. En cas de Raynaud sévère et réfractaire, un traitement par iloprost peut être proposé. En prévention secondaire des ulcères digitaux, le bosentan peut avoir un intérêt par analogie chez l’adulte. En cas d’atteinte pulmonaire interstitielle, un traitement par cyclophosphamide était jusqu’à récemment le traitement de référence, mais plusieurs publications ont montré une efficacité avec moins d’effets secondaires, du mycophénolate mofétil. Le traitement de l’atteinte digestive est symptomatique (inhibiteur de la pompe à protons, prokinétique, etc.).
En cas d’atteinte cardiaque ou d’HTAP, une prise en charge par des équipes spécialisées est nécessaire. Dans les cas les plus graves, une greffe de cellules-souches hématopoïétiques a été proposée. En association au traitement médicamenteux, une prise en charge en kinésithérapie est indispensable, afin d’assouplir la peau à l’aide de massages, d’entretenir les amplitudes articulaires, et de lutter contre la raideur articulaire. Le pronostic de la sclérodermie systémique chez l’enfant est meilleur que chez l’adulte avec une survie à 5 ans de 89 %. La maladie évolue selon 2 groupes : soit de façon lente et insidieuse avec une faible mortalité, soit avec une atteinte d’organe sévère évoluant rapidement vers le décès en moins de 5 ans. Les facteurs prédictifs de mortalité au diagnostic chez l’enfant sont la fibrose pulmonaire (OR : 11,2), l’élévation du taux de créatinine (OR : 22,7) et la péricardite (OR : 41,3)(10).
Conclusion
On retiendra que la sclérodermie localisée juvénile est une maladie à connaître chez l’enfant pouvant entraîner des séquelles fonctionnelles et esthétiques. La sclérodermie systémique juvénile est rare, mais peut être sévère avec des atteintes d’organes vitaux, nécessitant une prise en charge en centre spécialisé.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :