Publié le 05 déc 2022Lecture 6 min
Trouble héréditaire de la conduction cardiaque
Sonia SALMI-BELMIHOUB, Philippe CHEVALIER, Service de rythmologie, hôpital cardiologique Louis Pradel, Lyon
La rubrique « En direct des staffs » est ouverte à tout médecin d’un service de pédiatrie souhaitant partager avec les lecteurs de Pédiatrie Pratique les cas discutés dans son service et qu’il estime suffisamment intéressants et édifiants pour être portés à la connaissance de ses confrères.
La maladie progressive du tissu de conduction cardiaque (PCCD : progressive cardiac conduction disease) est un trouble héréditaire responsable d’un processus dégénératif du tissu de conduction cardiaque, il peut être isolé ou associé à une cardiopathie congénitale et peut progresser jusqu’au bloc atrio-ventriculaire complet (BAVC).
Nous rapportons le cas de P. L. né le 26 janvier 2017 appareillé d’un pacemaker simple chambre épicardique à l’âge de 17 mois pour bloc atrio-ventriculaire de haut degré. Il s’agit d’un enfant au développement staturopondéral normal, asymptomatique chez qui l’on découvre à l’âge de 7 mois lors d’une hospitalisation pour gastroentérite des troubles de la conduction : BAV 2/1 intermittent, bloc de branche droit incomplet avec une durée du QRS à 117 ms, un espace PR limite à 197 ms.
L’échographie cardiaque montre une architecture cardiaque normale, il n’y a pas de communication interauriculaire ou interventriculaire, l’arche aortique est normale. L’ECG réalisé à la mère est normal, l’ECG du père objective une bradycardie sinusale à 54/min, un espace PR à 160 ms et un BBDI avec une durée des QRS à 105 ms.
Il n’y a pas d’anticorps anti-SSA et anti-SSB chez la mère, et pas d’antécédents de cardiopathie congénitale dans la famille.
Une étude génétique met en évidence des variations sur l’exon 17 du gène TRPM4, ce variant a été associé à des anomalies de la conduction cardiaque(8).
P. L. est régulièrement surveillé par Holter rythmique qui montre une baisse progressive de la fréquence cardiaque moyenne sur 24 heures à 50 battements par minute, ainsi qu’une aggravation des troubles conductifs avec de nombreux passages en bloc atrio-ventriculaire du troisième degré, l’échographie cardiaque objective un début de retentissement du trouble conductif avec dilatation du ventricule gauche (figures 1 à 3).
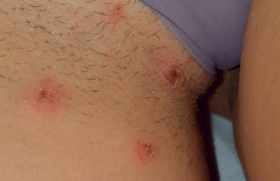
Figure 1. ECG de l’enfant.
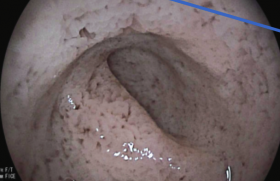
Figure 2. ECG de la mère.
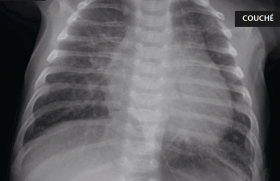
Figure 3. ECG du père.
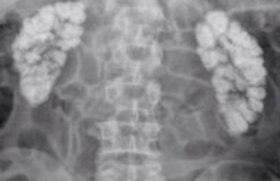
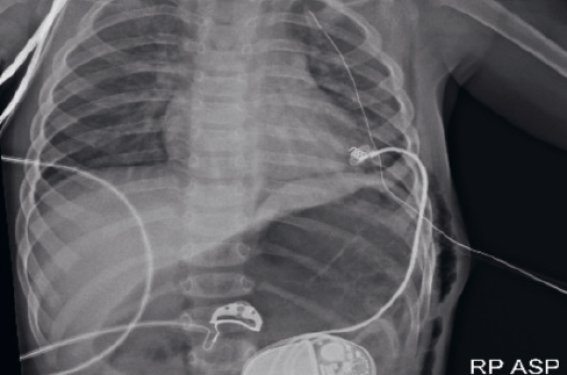
L’indication de stimulation cardiaque est retenue et il est procédé à la mise en place d’un stimulateur cardiaque simple chambre par voie épicardique avec sonde ventriculaire implantée à la pointe du ventricule gauche par thoracotomie gauche ; le boîtier est mis en place par voie pararectale gauche (figure 4).
Figure 4. Radiographie de thorax après implantation du P. M.
L’évolution a été marquée par une élévation modérée du seuil de stimulation ventriculaire à trois mois de l’implantation qui s’est par la suite stabilisée.
Étant donné une bonne accélération spontanée à l’effort et l’absence de signes fonctionnels, le stimulateur cardiaque est réglé en mode VVI sentinelle à 50/min et P. L. est stimulé dans 44 % du temps. Nous avons vu en consultation le pourcentage de stimulation augmenter avec le temps. Le contrôle échocardiographique montre un retour à la normale du ventricule gauche qui n’est pas dilaté avec une fonction ventriculaire gauche systolique de bonne qualité.
Discussion
Les troubles conductifs de l’enfant peuvent survenir sur un coeur structurellement normal ou associé à une cardiopathie congénitale. Ils peuvent aussi se voir dans les suites d’une chirurgie cardiaque.
Le bloc atrio-ventriculaire est dit congénital lorsqu’il est diagnostiqué in utero, à la naissance ou dans le premier mois de vie. Sa prévalence est 1 sur 15 000 à 20 000 naissances. Une maladie maternelle auto-immune représente 90 % des cas de bloc atrio-ventriculaire diagnostiqué avant l’âge de 6 mois(2), le passage de la barrière placentaire des auto anticorps anti-RO/SSA et anti-LA/SSB entraîne une fibrose irréversible du tissu de conduction foetal.£
Dans les autres cas où aucune cause évidente de bloc atrio-ventriculaire n’a été identifiée, le bloc cardiaque est dit idiopathique.
Dans une étude à grande échelle(1,2) sur une population pédiatrique avec BAV idiopathique, Baruteau et coll. ont observé un haut degré de caractère héréditaire et une participation génétique importante dans la pathogénie des BAV congénitaux et BAV isolés non auto-immuns de l’enfant, environ 70 % des enfants. avec bloc incomplet cardiaque ont progressé vers un bloc complet permanent suggérant un processus dégénératif post-natal du tissu de conduction.
Le PCCD ou trouble progressif de la conduction cardiaque est un trouble héréditaire caractérisé par un retard de la conduction cardiaque à travers le système His-Purkinje avec bloc de branche droit ou gauche pouvant évoluer vers un bloc atrio-ventriculaire complet (BAVC), syncope, voire mort subite. Il peut être isolé ou associé à une cardiopathie structurelle.
Il s’agit d’une dégénérescence et d’une fibrose progressive du tissu de conduction.
Les développements de la biologie moléculaire et de la génétique ont permis la découverte des bases génétiques de certaines formes familiales de PCCD.
En l’absence de cardiopathie structurelle congénitale ou de malformation systémique, le PCCD familial résulte de mutations de gènes codant des canaux ioniques impliqués dans la conduction de l’influx électrique, alors que le PCCD avec cardiopathie structurelle est dû à des mutations de gènes codant des facteurs de transcription, des enzymes et des protéines de structure(3).
Les mutations génétiques causales de la maladie dans le cas de PCCD avec coeur structurellement normal ont été identifiées dans les gènes codant pour les canaux ioniques SCN5A, SCN1B, SCN1OA, TRPM4 (cas de P. L.) et KCNK17.
Les porteurs de la mutation SCN5A, canalopathie sodique peuvent présenter des manifestations de syndrome du QT long type 3, de syndrome de Brugada associé à une atteinte de la conduction cardiaque.
Des mutations dans les gènes NKX2, TBX5, PRKAG2 et LMNA, sont responsables de PCCD associé à une cardiopathie congénitale.
La mutation NKX2.5 est à l’origine de PCCD associé à différentes cardiopathies congénitales tel que la communication interauriculaire ostium secundum(7), la tétralogie de Fallot, truncus artériosus, la transposition des gros vaisseaux, l’interruption de l’arche aortique.
Un certain nombre de PCCD avec test génétique négatif pour les gènes actuellement reconnus suggèrent que seule une partie de ces gènes défectueux a été identifiée.
Le diagnostic de PCCD familial isolé est suspecté chez des patients de moins de 50 ans avec troubles conductifs inexpliqués, coeur structurellement normal et des antécédents familiaux de syncope, stimulation cardiaque ou mort subite. Sa transmission est autosomale dominante avec une pénétrance variable(1).
L’évolution du PCCD peut se compliquer de dilatation ventriculaire gauche, insuffisance cardiaque, torsade de pointe et mort subite. Le risque de développer une myocardiopathie dilatée est important.
La fièvre étant un facteur déclencheur aggravant la maladie chez les porteurs de mutation du gène SCN5A, elle doit être traitée de manière préventive.
Une incidence élevée de mort subite est observée chez les patients présentant un bloc atrio-ventriculaire du premier degré associé à un bloc bifasciculaire ou chez les patients présentant un bloc atrio-ventriculaire avancé symptomatique.
Le traitement du PCCD familial est l’implantation d’un stimulateur cardiaque permanent.
Un suivi des patients est recommandé tous les six mois, quel que soit le degré du bloc cardiaque.
L’examen clinique des membres de la famille au premier degré est nécessaire lorsque le diagnostic du PCCD familial est établi chez un cas index et un examen annuel est recommandé pour les membres de la famille concernée présentant un ECG normal.
Non appareillé, le bloc atrio-ventriculaire congénital est associé à une mortalité estimée entre 14 et 34 %(1).
Il n’y a pas pour le moment de stratification du risque basée sur le génotype pour les patients atteints de PCCD.
La Heart Rythm Society (HRS) recommande l’implantation d’un pacemaker en cas de PCCD avec BAV 3 intermittent ou permanent, BAV de haut degré, ou BAV du 2e degré type I ou II symptomatique (classe I).
La stimulation est utile (classe IIa) en cas de bloc bifasciculaire avec ou sans BAV I.
Le pronostic est excellent chez les patients implantés d’un pacemaker sauf en présence de patients porteurs de mutation LMNA, SCN5A, où un risque de mort subite par trouble rythmique ventriculaire persiste, c’est pourquoi, la mise en place d’un défibrillateur doit être discutée(3).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :