Publié le 31 aoû 2020Lecture 7 min
Soif intense et énurésie secondaire
Bertrand CHEVALLIER, Service de pédiatrie, hôpital Ambroise-Paré, Boulogne-Billancourt

La rubrique « En direct des staffs » est ouverte à tout médecin d’un service de pédiatrie souhaitant partager avec les lecteurs de Pédiatrie Pratique les cas discutés dans son service et qu’il estime suffisamment intéressants et édifiants pour être portés à la connaissance de ses confrères.
Scène 1 - Un tableau clinique évocateur
Tom est adressé dans le service de pédiatrie à l’âge de 4 ans en raison d’une polyurie diurne et nocturne et une prise de boissons plus abondante, constatée par les parents depuis environ 2 mois. Il mesure 99 cm pour un poids de 14,8 kg. Sa vitesse de croissance staturale est globalement régulière avec une stagnation depuis la mesure chez son pédiatre, trois mois auparavant. Ses antécédents personnels et familiaux ne révèlent rien. Le développement psychomoteur est normal. L’examen clinique général ne montre aucune anomalie patente ; ses organes génitaux sont normaux pour son âge. Le syndrome polyuropolydipsique de Tom est authentifié par la réapparition récente d’une énurésie nocturne et d’une polyurie > 100-110 ml/kg/j. Les polyuries osmotiques (glycosurie et hypercalciurie) sont éliminées par la mesure d’une glycémie (0,75 g/l) et d’une calcémie (2,4 mmol/l). Les premières 24 heures d’observation dans le service confirment le diabète insipide, avec une diurèse mesurée à 2,5 litres, une osmolarité urinaire basse et la constatation d’une énurésie nocturne.
L’épreuve de restriction hydrique n’est pas effectuée en raison de sa possible dangerosité dans ce contexte de diurèse très importante. La potomanie étant peu probable. La natrémie est à 147mmol/l. La fonction rénale est normale.
Le test au Minirin® (deux pulvérisations de 10 μg pour cet enfant de plus de 10 kg) confirme l’origine centrale de son diabète insipide en entraînant une réduction du volume urinaire qui passe de 410 ml dans les 3 heures précédentes à 35 ml dans les 3 heures suivantes, avec une augmentation simultanée de l’osmolarité urinaire de 300 à 950 mOsmol/kg. La concentration d’ADH de base est effondrée.
Un diabète insipide d’origine centrale est ainsi affirmé.
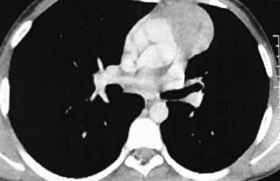
Une IRM cérébrale, centrée sur la région hypophysaire et le 3e ventricule, est réalisée à la recherche de signes pouvant évoquer une tumeur (craniopharyngiome ou dysgerminome). Elle montre une augmentation d’épaisseur isolée de la tige pituitaire avec une disparition de l’hypersignal de la posthypophyse. Il n’y a pas de processus expansif intra- ou suprasellaire. La région pinéale est normale.
La normalité des dosages des hormones sécrétées par l’antéhypophyse (FSH, LH, ACTH, TSH), de l’IGF1 et du cortisol plaident pour un diabète insipide central isolé. Les marqueurs pouvant faire évoquer une tumeur germinale (a-foetoprotéine, ß-hCG) sont normaux. On complète le bilan étiologique par un examen ORL spécialisé, une scintigraphie osseuse et des radiographies du crâne, dans le cadre d’une possible histiocytose langerhansienne : ces examens sont normaux. La normalité de tous ces examens n’exclut en rien un diagnostic à venir toujours possible de dysgerminome ou d’histiocytose car l’imagerie, dans ces situations, peut être retardée de plusieurs mois ou années. Une nouvelle imagerie cérébrale est programmée dans les six mois suivants.
À ce stade : un diabète insipide isolé, sans cause identifiée.
La famille déménageant pour vivre à l’étranger (Canada), les éléments du suivi ultérieur sont imprécis. On sait simplement que le traitement par desmopressine (Minirin®) par voie endonasale en deux prises quotidiennes a été efficace, avec une disparition quasi-complète des symptômes urinaires et de la soif excessive. L’IRM cérébrale, réalisée neuf mois plus tard, est superposable à l’imagerie initiale.
Scène 2 - Tom à l’âge de 7 ans
Figure 1. Ralentissement de la vitesse de croissance staturale entre 5 et 7 ans.
Tom inquiète sa famille, car il ne grandit pas bien. Il mesure en effet 107 cm à l’âge de 7 ans et n’a pris que 2 cm en 15 mois (figure 1). Les examens biologiques vont rapidement confirmer l’hypothèse première des endocrino-pédiatres canadiens qui suivent l’enfant. Le test de stimulation de l’hypophyse sous l’association glucagon-bétaxolol ne montre qu’un pic de 11 UI/ml (5,5 ng/ml) d’hormone de croissance (GH), avec une concentration d’IGF1 à - 2,5 DS pour son âge. Une sécrétion diminuée de GH est confirmée par une réponse insuffisante (6,7 ng/ml) lors d’un second test de stimulation avec hypoglycémie insulinique.
Figure 2. IRM : épaississement de la tige pituitaire et disparition de l’hypersignal de la posthypophyse.
Le diagnostic de déficit en hormone de croissance est établi. Les autres lignées hypophysaires sont normales. L’IRM est identique avec une majoration possible de l’épaississement de la tige pituitaire et une absence de l’hypersignal de la posthypophyse (figure 2). Un nouveau bilan radiologique est effectué. Il montre une lacune osseuse dans la région pariétale gauche : le reste du bilan osseux et ORL est normal.
La découverte d’une lacune osseuse sur la radiographie du crâne retient le diagnostic probable d’histiocytose langerhansienne (HL) dans sa forme localisée
• L’HL est une prolifération de cellules de Langerhans anormales au sein de divers tissus. Le diagnostic repose sur l’examen histologique d’un tissu atteint lorsqu’il est accessible.
• L’HL regroupe plusieurs entités cliniques anciennement dénommées granulome éosinophile, maladie de Hand-Schuller-Christian, maladie de Letterer-Siwe et maladie d’Hashimoto-Pritzker. Il y a une accumulation d’histiocytes (macrophages) présentant des marqueurs spécifiques des cellules de Langerhans (CD1a, Langerine et granule de Birbeck) dans les tissus atteints. Avant 15 ans, l’incidence de cette maladie est estimée à 5 cas par million, soit environ 60 nouveaux cas par an en France. L’âge moyen au diagnostic est de 3,5 ans. La biopsie cutanée en cas de lésion est la plus contributive.
• L’HL se présente sous deux formes : l’atteinte d’un seul organe (principalement l’os ou la peau) ou l’atteinte multisystémique à rechercher systématiquement quand le diagnostic est évoqué.
L’atteinte osseuse peut être isolée ou multiple. Les principales localisations sont les os du crâne, la mastoïde, la diaphyse des os longs et les vertèbres. Ces lésions se présentent comme une lyse osseuse à l’emporte-pièce associées à une soufflure de la corticale et/ou à une réaction périostée, et au niveau des vertèbres par des tassements vertébraux (vertebra plana). La lyse de la mastoïde peut se manifester par une otite moyenne chronique et une otorrhée chronique, correspondant à une infiltration du conduit auditif externe. L’atteinte labyrinthique entraîne des signes fonctionnels comme un vertige ou une surdité aigüe.
L’atteinte cutanée est polymorphe. Les lésions sont le plus souvent papulo-squameuses plus ou moins pétéchiales touchant principalement le cuir chevelu (toujours y penser lors de l’examen clinique), la face antérieure du tronc et les plis axillaires ou inguinaux pouvant prêter à confusion avec un eczéma ou une dermatite séborrhéique.
L’atteinte hématologique est une anémie ou une thrombopénie, rarement une neutropénie, induite par l’infiltration de la moelle osseuse par les cellules de Langerhans et de mauvais pronostic. Le bilan est alors complété par un myélogramme, voire une biopsie médullaire.
L’atteinte hépatique se présente sous deux formes :
– aiguë, avec une hépatomégalie, une cytolyse, voire une insuffisance hépatocellulaire d’évolution favorable avec un traitement adapté ;
– chronique, avec une cholangite sclérosante, responsable d’une cirrhose biliaire d’évolution irréversible.
• Le diabète insipide peut précéder (16 %), accompagner (28 %), ou succéder (56 %) aux autres localisations de la maladie, et en particulier à une infiltration de l’antéhypophyse, responsable d’une insuffisance en GH. Il faut donc s’attacher à rechercher des atteintes périphériques biopsiables (lésion cutanée ou osseuse périphérique). Le délai moyen entre le diagnostic d’un diabète insipide isolé et l’apparition d’une seconde localisation d’histiocytose est en moyenne de 1,3 an.
Messages à retenir concernant le diabète insipide central de l’enfant
• Un syndrome polyuro-polydipsique de l’enfant doit avant tout faire évoquer un diabète sucré.
• En l’absence de glysosurie, évoquer un diabète insipide après inogramme sanguin, mesure de l’osmolarité sanguine et/ou urinaire.
• Le test de restriction hydrique différencie la polyurie primaire (potomanie) ou secondaire ( diabète insipide) : il doit être pratiqué en milieu hospitalier.
• L’épreuve au Minirin® différencie le diabète insipide central (ascension de l’osmolarité urinaire > 600 mosm) ou périphérique : insensibilité rénale à l’ADH (l’osmolarité urinaire reste basse).
• Un diabète insipide central indique une IRM cérébrale en urgence (recherche de tumeurs).
• L’instauration d’un traitement par Minirin® doit être encadrée (importance de l’éducation thérapeutique) pour éviter une hyponatrémie et une inflation hydrique.
• Une IRM cérébrale doit être répétée un an après, voire au bout de 3-5 ans : signes retardés à l’imagerie.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :