Publié le 12 fév 2008Lecture 7 min
Un gros foie fébrile à 4 mois
D. ARMENGAUD, CHI, Poissy/Saint-Germain-en-Laye
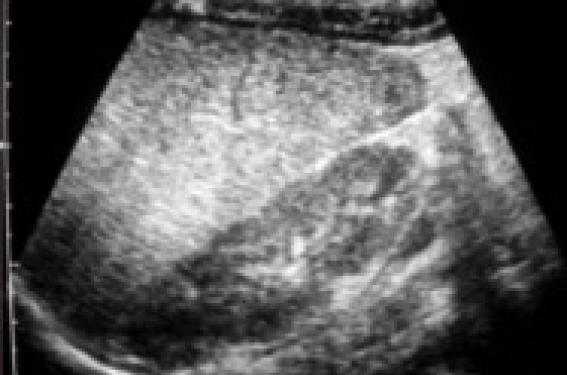
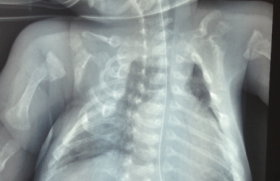
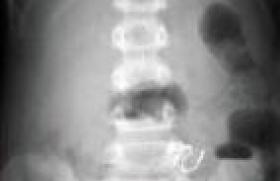
Histoire clinique À la consultation, le médecin retrouve une rhinopharyngite avec une rhinite mucopurulente postérieure et des tympans qui paraissent infiltrés, dépolis sans collection purulente sous-jacente. Cela semble expliquer la fièvre et l’enfant ne présente pas de signes respiratoires préoccupants… cependant, l’examen clinique retrouve une hépatomégalie importante et un pôle de rate palpable, signes qui, d’après les parents, n’étaient pas connus jusqu’à présent. A gauche, figure 1 : ASP. Hépatomégalie et splénomégalie. Au centre, figure 2 :Radiographie pulmonaire normale . A droite, figure 3 : Échographie hépatique : hépatomégalie hétérogène hyperéchogène (par rapport au rein), sans syndrome tumoral. Examens biologiques • NFS : – Hb : 8,6 g/100 ml – GR : 2 950 000/mm3 (VGM 88 µ3) – GB : 9 800/mm3 (dont 81 % de PNN) – Plaquettes : 191 000/mm3 • Glycémie : 5,9 mmol/l • Ionogramme sanguin : (mmol/l) : Na : 140 ; K : 5,0 ; bicarbonates : 22 ; Ca : 2,25 ; urée : 4,1 • Protides : 62 g/l • Créatinine : 34 µmol/l • Bilirubine : 4 µmol/l L’enfant est né à terme, après une grossesse sans particularité, avec un poids de naissance de 3 100 g, pour une taille de 49,5 cm et un PC de 35 cm. Il a présenté un ictère néonatal durant quelques semaines, ce qui a semblé banal à la mère ayant eu au cours de ses deux grossesses, la précédente et celle de Bryan, un ictère gravidique avec prurit. Bryan est adressé aux urgences en raison de cette hépatomégalie inconnue et de découverte « fortuite ». À l’examen, l’enfant paraît en bon état général, l’hémodynamique est correcte (fréquence cardiaque à 98/min, pression artérielle à 85/56 mmHg) pour une température de 37,8 °C, avec un temps de recoloration inférieur à 2 secondes. Il pèse 5 700 g (-1DS) pour une taille de 60 cm (M). L’auscultation pulmonaire est normale. La palpation de l’abdomen retrouve une hépatomégalie avec un débord de 11,5 cm atteignant l’ombilic sur la ligne mamelonnaire, de consistance dure avec un bord ferme. La splénomégalie déborde de 2-3 cm le rebord costal. Il n’y a pas d’ictère, ni d’épanchement séreux, ni prurit. L’examen ORL est normal ; il existe cependant une candidose buccale traînante. Un bilan biologique est prélevé et une échographie abdominale demandée. Quelle est votre démarche diagnostique et quels examens complémentaires demander pour l'étayer ? Histoire clinique (suite) La découverte surprenante de cette hépatomégalie jusque-là méconnue pose la question d’une hépatomégalie tumorale à progression rapide, et donc, à cet âge, en premier lieu à : • un syndrome de Pepper, forme particulière du neuroblastome ; mais la consistance du foie, la présence d’une splénomégalie, et… paradoxalement, la perturbation du bilan hépatique ne vont pas dans ce sens. L’échographie n’apporte pas d’éléments suffisamment fiables dans ce contexte pour éliminer formellement une tumeur surrénalienne parfois déjà disparue ; la recherche d’une augmentation des catécholamines urinaires s’impose : les résultats reviendront normaux ; • un hépatoblastome, de croissance plus lente ; mais l’aspect échographique (absence de tumeur individualisée, absence d’amputation du système porte) et le dosage demandé de l’alpha fœtoprotéine très modérément élevée à 55 ng/ml (< 6,05) permettent, en pratique, de l’éliminer. Un examen TDM est demandé. Il confirme l’hépatomégalie hétérogène sans nodule individua-lisable. La vésicule et les voies biliaires sont normales. L’injection ne montre pas d’amputation du système porte (figures 4 A et B). Figure 4. (A) ( à gauche) TDM sans injection. Hépatomégalie hétérogène, sans nodule individualisable. (B) (à droite) TDM avec injection. Absence de prise de contraste anormale, pas d’amputation du système porte. Il faut donc plutôt s’orienter vers une « hépatopathie » d’évolution précoce, ce d’autant que l’on retrouve la notion d’un ictère prolongé qui, chez cet enfant né et en absence de toute histoire d’hémolyse et d’allaitement maternel, devrait être probablement cholestatique ; encore eut-il fallu le préciser ! Rappelons une fois de plus la nécessité d’explorer et de « qualifier » tout ictère. Les caractéristiques cliniques de l’hépatomégalie, associées aux anomalies biologiques (élévation des transaminases et des enzymes de cholestase), peuvent faire évoquer : • une atrésie des voies biliaires ; mais à cet âge et sans ictère clinique, cela ne peut être retenu, et la présence d’une vésicule biliaire en échographie, sans lithiase ni dilatation des voies biliaires, élimine une lithiase ou un kyste du cholédoque ; • une mucoviscidose, malgré le dépistage systématique, qui cependant n’est pas exhaustif, car ne repérant que les mutations les plus fréquentes. Le test de la sueur est normal (12 mmol/l ; N < 60) et un dosage de la trypsine immuno-réactive (13,3 µg/l ; N < 60) ne vont pas dans ce sens ; • un syndrome d’Alagille ; mais le poids de naissance est normal, il n’y a pas de dysmorphie faciale, pas de souffle cardiaque ni de vertèbre en aile de papillon sur la radiographie du thorax ; • une maladie de surcharge type Niemman-Pick ; mais la taille de la splénomégalie est en général bien supérieure à l’hépatomégalie ; • un déficit en alpha 1 antitrypsine, mais l’électrophorèse des protéines est normale : – albumine : 37,7 g/l (37-47) – alpha 1 globulines : 1,5 g/l (1-3) – alpha 2 globulines : 1 g/l (9-11) – gamma globulines : 3,3 g/l (7-14). Avant d’envisager une exploration invasive (biopsie hépatique ou cholangiographie), n’y a-t-il pas une vérification à faire ? car le diagnostic est là ! Si l’électrophorèse des protides est normale dans son résultat brut, c’est le tracé électrophorétique… (qui n’est pas toujours adressé) qui montre une absence de pic d’alpha-1 (figures 5) évocateur d’un déficit en alpha-1 antitrypsine (AAT) ; il sera confirmé par : – le dosage spécifique à 0,54 g/l (1,03-2,16) ; – l’identification de la mutation : homozygotie ZZ chez cet enfant. Figure 5. Électrophorèse des protides : absence de pic d’alpha 1 globuline. Commentaire L’alpha-1 antitrypsine (ATT), principale molécule inhibitrice des protéases (Pi) est une glycoprotéine codée par un gène situé sur le chromosome 14 présentant plusieurs allèles : normal (M), déficient (Z, S) ou absent (nul). La mutation Z (glu342lys) est portée par 4 % de la population et seul l’état homozygote (ZZ ou plus rarement nul-nul) ou de double hétérozygote (Z-nul) conduit à : – une cholestase néonatale transitoire dans 10 % des cas ; – une évolution vers la cirrhose chez 10 % de ces enfants, dont l’évolution faite de cholestase chronique, d’hypertension portale et d’insuffisance hépato-cellulaire mène à envisager une transplantation hépatique. • Chez l’enfant, le déficit en AAT se révèle principalement par une atteinte hépatique liée à l’accumulation intracellulaire dans le réticulum endoplasmique de cette molécule mutée, qui est identifiable en histologie sous forme de granulations PAS +. • La mutation de cette protéine est à l’origine d’une modification de sa configuration spatiale, dont le site actif se fixe alors préférentiellement non pas sur sa cible habituelle, une protéase, mais sur une autre molécule d’AAT ; cela conduit à une polymérisation et ainsi à son accumulation dans le réticulum endoplasmique, qui ne peut la dégrader. Le nouveau-né serait particulièrement exposé par l’altération de cette protéolyse cellulaire. Cette dernière peut être majorée à l’occasion d’une infection intercurrente par augmentation de la synthèse protéique hépatique, et de la fièvre qui augmenterait la polymérisation. D’autres facteurs interviennent peut-être car tous les porteurs du génotype ZZ ne font pas pour autant une maladie hépatique. • Chez l’adulte, le déficit plasmatique en AAT est à l’origine d’un défaut de la balance protéase-antiprotéase (l’AAT, est une anti-élastase, elle s’oppose aux élastases produites par les polynucléaires) ; il aboutit à la destruction des structures élastiques du poumon se traduisant par un emphysème souvent aggravé par des facteurs environnementaux (particulièrement le tabagisme),
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :