Publié le 10 avr 2011Lecture 13 min
Pièges cliniques en rhumatologie chez l’enfant
P. QUARTIER, Université Paris-Descartes et Centre national de référence des maladies rares « Arthrites juvéniles », Unité d’imm
Partant de cas cliniques bien réels et parfois un peu simplifiés, nous souhaitons illustrer ici l’importance d’une démarche d’analyse clinique prudente pour ne pas passer à côté d’un diagnostic, dont une prise en charge inadaptée pourrait avoir des conséquences sur le pronostic vital ou fonctionnel du jeune patient.
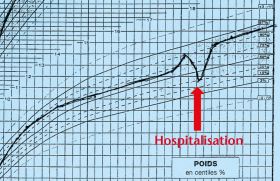
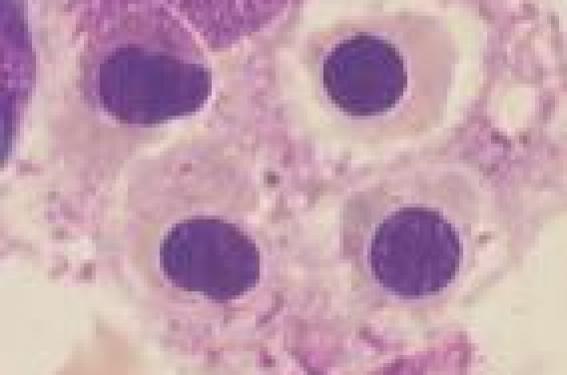
La rhumatologie pédiatrique couvre un ensemble de maladies inflammatoires de l’enfant à expression systémique et/ou articulaire que l’on peut répartir en quatre grandes catégories : les arthrites juvéniles idiopathiques (AJI), caractérisées par une arthrite débutant avant l’âge de 16 ans, durant plus de 6 semaines et de cause inconnue ; les maladies de système auto-immunes et les vascularites à début pédiatrique ; les syndromes et maladies auto-inflammatoires, dont certaines granulomatoses ; des atteintes de l’appareil locomoteur n’entrant dans aucune catégorie ci-dessus, ni dans le champ de maladies génétiques, malformatives, infectieuses ou oncologiques. Dans cette catégorie figurent les arthrites réactionnelles et des maladies psychosomatiques, dont certaines algodystrophies et autres fibromyalgies de l’enfant. Le diagnostic positif et différentiel des maladies rhumatologiques pédiatriques est essentiellement clinique, avec de nombreux pièges liés au caractère souvent peu spécifique des symptômes et à la fréquence de présentations atypiques. Situation 1. Boiterie et épanchement du genou Un enfant est amené par son père à la consultation pour l’apparition d’un gros genou à droite entraînant une boiterie. Le père est suivi pour une spondylarthrite ankylosante. L’interrogatoire relève un épisode d’allure virale 3 semaines auparavant, un début des symptômes depuis 4 jours. Des examens complémentaires réalisés en ville 48 heures avant l’actuelle consultation mettent en évidence une hyperleucocytose à polynucléaires neutrophiles, une élévation de la vitesse de sédimentation (VS) et de la protéine C-réactive (CRP), un typage HLA B27 positif comme chez le père de l’enfant et des radiographies de genou normales. ● Pièges à éviter : évoquer une arthrite juvénile idiopathique débutante (éventuellement spondylarthropathie juvénile eu égard au contexte familial et à la positivité du HLA B27, sachant que le mode d’entrée peut être une monoarthrite du genou) ou une arthrite réactionnelle (épisode d’allure virale un peu plus de 2 semaines avant le début des symptômes), alors qu’il s’agit de diagnostics d’élimination. ● Urgence diagnostique et thérapeutique : craindre une arthrite septique devant toute monoarthrite associée à une fièvre ou une biologie inflammatoire. Il faut réaliser une ponction du liquide articulaire de l’épanchement du genou à visée cytologique et bactériologique, avec un lavage articulaire en cas de liquide épais purulent et effectuer des prélèvements d’une éventuelle porte d’entrée cutanée et une hémoculture, et débuter une bithérapie antibiotique IV associée à une immobilisation de l’articulation par plâtre prenant les articulations sus- et sous-jacente. Situation 2. Fièvre élevée, continue et mal tolérée depuis 6 jours chez un nourrisson de 7 mois Cette fièvre est associée à des arthralgies et à un rash cutané maculeux, avec un syndrome inflammatoire biologique marqué. Les recherches microbiologiques sont négatives. Une antibiothérapie à large spectre a été débutée au second jour de la fièvre, sans effet. ● Pièges à éviter : – multiplier les examens recherchant une cause infectieuse, alors même que l’hypothèse d’une infection bactérienne est peu probable (antibiothérapie à large spectre inefficace) en méconnaissant la possibilité d’un diagnostic alternatif urgent ; – évoquer à tort une forme systémique d’arthrite juvénile idiopathique (maladie de Still de l’enfant) sur l’association fièvre-éruption- arthralgies, alors même que le très jeune âge de l’enfant rend cette hypothèse peu probable et que le caractère continu de la fièvre s’oppose à la courbe thermique de la maladie de Still (pics hyperthermiques entrecoupés de franches défervescences thermiques). ● Urgence diagnostique et thérapeutique : évoquer systématiquement une maladie de Kawasaki devant une fièvre de plus de 5 jours chez un enfant de moins de 5 ans, même en l’absence de l’ensemble des signes cliniques typiques, dont, notamment, la desquamation cutanée tardive (rechercher cependant une chéléite parfois a minima avec des lèvres discrètement fissurées, plus spécifique que la pharyngite, la conjonctivite ou les adénopathies). Il faut hospitaliser l’enfant et demander une échographie cardiaque à la recherche d’une atteinte coronarienne anévrysmale (noter le risque accru d’anévrysmes géants chez le petit nourrisson). Le traitement repose sur les immunoglobulines intraveineuses, l’aspirine à dose antiinflammatoire et parfois l’héparinothérapie (si anévrysme volumineux). Situation 3. Arthrites aseptiques à bascule chez un patient avec un déficit immunitaire La maladie type est la maladie de Bruton (agammaglobulinémie liée à l’X). Les patients bénéficient d’une substitution mensuelle en immunoglobulines. Ces arthrites évoluant sur plusieurs semaines sont associées à une fébricule intermittente et à des céphalées croissantes chez un jeune garçon avec un fléchissement des performances scolaires depuis 1 an. ● Pièges à éviter : ne pas prendre en compte le terrain et omettre de prendre avis auprès d’un médecin suivant régulièrement des patients avec déficit immunitaire. ● Diagnostic : doivent être pris en compte le déficit immunitaire sous-jacent et une substitution en immunoglobulines probablement insuffisante. Sur une agammaglobulinémie, la demi-vie des immunoglobulines perfusées impose une substitution au minimum toutes les 3 semaines. La chronicité de l’atteinte, le caractère aseptique des arthrites et l’association d’une touche neurologique (céphalées, fléchissement scolaire) plaident pour une infection chronique à entérovirus, seule famille de virus à l’origine d’infections sévères dans des déficits purs de l’immunité humorale, pouvant donner une méningo-encéphalite chronique (recherche par PCR de l’entérovirus dans le LCR), des arthrites aseptiques, une myocardite (échovirus = famille des entéro-virus), une atteinte cutanée et myositique. Situation 4. Fièvre devenant continue et mal tolérée chez un patient traité pour une forme systémique d’arthrite juvénile idiopathique C’est ce que l’on peut observer en cas de maladie de Still pédiatrique après introduction d’un traitement par indométacine. ● Pièges à éviter : évoquer un problème infectieux, mettre en oeuvre une antibiothérapie sans modifier le traitement anti-inflammatoire. ● Diagnostic : il s’agit d’un syndrome d’activation macrophagique (SAM) (figure 1) venant compliquer une forme systémique d’arthrite juvénile idiopathique. Figure 1. Hémophagocytose au cours du syndrome d’activation macrophagique (observation n°4). Il faut réaliser les examens biologiques appropriés (tableau 1), hospitaliser l’enfant, arrêter le toxique potentiel (en l’occurrence l’indométacine, par ailleurs très bon traitement de première ligne de cette maladie) et débuter en urgence une corticothérapie générale éventuellement associée à un traitement par ciclosporine. Tableau 1. Syndrome d’activation macrophagique (SAM). Circonstances favorisantes : • Le terrain : – certaines maladies inflammatoires dont la forme systémique d’arthrite juvénile idiopathique (FS-AJI) et le lupus érythémateux disséminé ; – maladies génétiques avec défaut de contrôle de l’activation lymphocytaire T (lymphohistiocytoses familiales, syndrome de Purtillo [rôle déclenchant essentiellement de l’EBV], syndromes de Chediak-Higashi et de Griscelli, etc.) ; – déficits immunitaires avec défaut de réponse aux agents intracellulaires ; – certaines hémopathies malignes (lymphomes T ou NK) ; – certaines maladies métaboliques (déficit en protéine dibasiques, Gaucher, etc.). • Les facteurs déclenchants : – agent infectieux intracellulaire (EBV, CMV, toxoplasmose, etc.) ; – toxiques en fonction du terrain (sur une FS-AJI, sels d’or et sulphasalazine : contre-indiqués car pourvoyeurs systématiques de SAM ; anti-inflammatoires non stéroïdiens et autres médications, plus rarement incriminés). Le diagnostic clinique et biologique : – fièvre élevée souvent mal tolérée et continue, parallèlement à une amélioration paradoxale des arthrites sur une FS-AJI, parfois adénomégalies, hépatosplénomégalies, signes hémorragiques, voire signes neurologiques (atteinte neuro-méningée essentiellement dans les déficits génétiques du contrôle de l’activation lymphocytaire T) ; – hyperferritinémie majeure, hypertriglycéridémie, cytopénie, coagulopathie avec fibrinogénopénie, altérations du bilan hépatique, etc. ; – excès majeur de lymphocytes T activés dans certaines maladies ; – parfois hémophagocytose objectivée sur un myélogramme ou un prélèvement biopsique. Quelques constantes de la prise en charge initiale : – hospitalisation (urgence thérapeutique) ; – traitement si possible du facteur déclenchant (anti-infectieux, arrêt d’un toxique) ; – corticothérapie à fortes doses (si possible précédée de prélèvements sanguins ± médullaires permettant d’avancer sur le diagnostic étiologique après avis spécialisé), éventuellement ciclosporine ; – avis spécialisé rapide en fonction de l’orientation étiologique. Situation 5. Convulsions chez une adolescente d’origine antillaise On retrouve à l’interrogatoire la notion de photosensibilité avec un discret rash malaire l’été précédent (figure 2). Il existe une discrète cytopénie sur la numérationformule sanguine. ● Pièges à éviter : méconnaître le diagnostic de lupus érythémateux disséminé et surtout méconnaître les causes possibles de convulsion sur ce terrain. ● Diagnostics à évoquer : voir le tableau 2. Figure 2. Érythème malaire lupique (observation n°5). Tableau 2. Symptômes neurologiques chez un patient atteint de lupus érythémateux disséminé. • Thrombose artérielle ou veineuse sur syndrome des antiphospholipides : – biologie : rechercher un anticoagulant circulant de type lupique, un allongement du TCA du témoin par le plasma du patient, anticorps anti-cardiolipine, voire un anti-bêta2 GP-1 ; – imagerie guidée par l’examen clinique (IRM ou scanner cérébral). • Hypertension artérielle « maligne » pouvant notamment survenir sur glomérulonéphrite lupique décompensée : importance de la surveillance régulière de la PA, de la bandelette urinaire et de la protéinurie des 24 h et de la fonction rénale. • Infection cérébro-méningée, a fortiori chez un patient sous immunosuppresseurs : – interrogatoire et examen clinique (porte d’entrée infectieuse, lésions herpétiques, etc.) ; – imagerie cérébrale, fond d’oeil, examens sanguins, virologie ; – discuter la ponction lombaire. • « Neurolupus », vascularite lupique : – après avoir éliminé l’une des causes précédentes ; – à craindre sur un LED mal suivi ou mal contrôlé, avec des signes cliniques et/ou biologiques de maladie lupique active ; – IRM cérébrale. • Toxicité des traitements : penser notamment à une mauvaise tolérance des corticostéroïdes sur un syndrome dépressif, après avoir éliminé une atteinte propre à la maladie lupique. Situation 6. Fièvre récurrente mensuelle depuis l’âge de 4 ans associée à une aphtose buccale importante et des adénopathies chez un garçon de 4 ans et demi Le tableau fait évoquer le diagnostic de syndrome de Marshall (ou PFAPA), avec des céphalées qui persistent et s’intensifient. ● Pièges à éviter : ne pas reconsidérer le diagnostic malgré les atypies (l’âge de début tardif au-delà de 2-3 ans, céphalées persistantes et intenses), et proposer une thérapeutique inappropriée (ici étaient envisagées une amygdalectomie et une adénoïdectomie sous anesthésie générale non dénuée de risque en la circonstance). ● Diagnostics : maladie de système (évoquer en second un processus infectieux régulièrement décapité, moins vraissemblablement une maladie maligne). L’association fièvre-aphtose céphalées intenses a conduit à réaliser un bilan sanguin montrant un syndrome inflammatoire, mais l’absence d’auto-anticorps, un examen ophtalmologique montrant une uvéite postérieure, une IRM cérébrale mettant en évidence une thrombose du sinus caverneux, conduisent au diagnostic de maladie de Behçet, vascularite inflammatoire avec aphtose volontiers bipolaire (mais pas toujours), uvéite postérieure, haut risque thrombogène chez le sujet jeune, parfois associée à un terrain génétique particulier (comme chez ce jeune garçon qui était HLA B51). Situation 7. Fièvre et myalgies intenses diffuses et continues depuis 2 semaines chez un jeune garçon d’origine turque ● Pièges à éviter : méconnaître cette présentation clinique d’une maladie fréquente dans la population turque, arménienne, juive et plus généralement du bassin méditerranéen, et se contenter d’antalgiques classiques, très peu efficaces en l’occurrence (autres présentations trompeuses : orchite, purpura vasculaire fébrile, lésions érysipéloïdes, etc.). ● Diagnostic : crise myalgique révélatrice d’une fièvre méditerranéenne familiale. Un avis spécialisé est impératif pour valider le traitement. Une approche thérapeutique encore novatrice, par l’antagoniste du récepteur de l’interleukine-1 (anakinra, Kineret ®), est à discuter au cas par cas et permet de stopper les symptômes en quelques heures, témoignant du rôle central de l’IL-1 dans ces maladies auto-inflammatoires. Ce traitement est à relayer par la colchicine pour prévenir la récurrence des crises et l’amylose secondaire. Situation 8. Syndrome inflammatoire prolongé inexpliqué chez une adolescente Le syndrome inflammatoire est secondairement associé à des douleurs à la marche de type claudication intermittente. ● Pièges à éviter : omettre la prise de pression artérielle si possible aux quatre membres et la palpation des pouls périphériques. ● Diagnostic : vascularite de Takayasu, 3e vascularite en fréquence dans le monde. Elle touche surtout la femme entre 10 et 50 ans. Elle est caractérisée par une fièvre ou parfois simplement un syndrome inflammatoire prolongé avec asymétries tensionnelle et asymétrie des pouls, voire abolition des pouls périphériques, développement progressif d’anévrysmes multiples et de sténoses sur l’aorte et ses branches. Le pronostic est très réservé en l’absence de prise en charge précoce (corticothérapie ± immunosuppresseurs, prise en charge cardiovasculaire). Conclusion D’autres situations cliniques justifient d’évoquer des maladies infectieuses mimant une maladie de système ou, à l’inverse, une maladie auto-inflammatoire mimant des infections récurrentes. Devant un début très précoce, penser au déficit en mévalonate kinase en congelant des urines en période de crise pour dosage de l’acide mévalonique urinaire et penser à la fièvre méditerranéenne familiale en fonction des origines ethniques des parents. D’autres pathologies peuvent encore être en cause, comme une myopathie d’origine génétique mimant une dermatomyosite juvénile, une maladie osseuse constitutionnelle en imposant pour une maladie inflammatoire de l’appareil locomoteur (tout en reconnaissant l’existence d’entités mixtes, dont certaines maladies génétiques associées à un lupus, des engelures, une petite taille et des anomalies osseuses particulières), une pathologie maligne avec des douleurs articulaires ou périarticulaires, une fragilité osseuse constitutionnelle ou encore une maltraitance d’enfant devant des fractures d’âges différents.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :