Publié le 15 jan 2008Lecture 15 min
Le généticien face au retard psychomoteur de l’enfant
D. LACOMBE, Service génétique médicale. Hôpital Pellegrin-Enfants, CHU de Bordeaux Laboratoire de génétique humaine. Université Victor-Segalen Bordeaux 2, Bordeaux
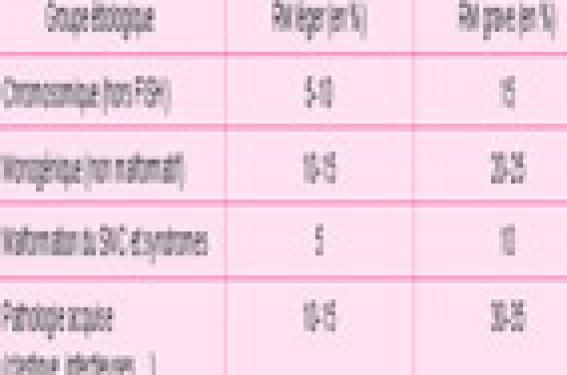
L’identification de la cause du retard psychomoteur de l’enfant est essentielle car elle seule permet de réponde aux questions de la famille sur l’étiologie, le pronostic, la prise en charge adaptée et le risque de récurrence. Les progrès, notamment dans l’identification des causes génétiques de retard mental, ont été importants ces dernières années, mais il reste encore trop de cas inexpliqués, imposant de poursuivre l’amélioration de nos connaissances dans ce domaine.
Le terme de retard psychomoteur est préféré à celui de retard mental chez les jeunes enfants avant 3 ans, en raison de l’impossibilité de séparer clairement le versant moteur du versant intellectuel. L’Association américaine sur le retard mental (AARM) définit le retard mental comme une diminution significative des facultés intellectuelles associée à un déficit des comportements adaptatifs survenus au cours de la période de dévelop-pement intellectuel(1). Les comportements adaptatifs sont les capacités de gérer les interactions sociales, la communication, les soins personnels, les compétences domestiques, les aptitudes sociales, l’utilisation des ressources communautaires, l’autonomie, la santé et la sécurité, les aptitudes en milieu scolaire, dans les loisirs et le travail. Épidémiologie Le niveau d’intelligence est évalué sous forme d’un quotient correspondant aux aptitudes comparées à une population de même âge chronologique, par des tests normés. Le quotient intellectuel (QI) exprime les capacités en référence à une échelle répartie de façon gaussienne dont la moyenne est de 100. On parle de retard mental vrai à partir d’un QI en dessous de 70. Le retard mental est qualifié de léger entre 50 et 70, de modéré entre 35 et 50, de sévère entre 20 et 35, et de profond en dessous de 20. La prévalence du retard psychomoteur est estimée entre 1 et 3 % chez les enfants de moins de 5 ans, soit 6 000 à 18 000 nouveaux cas par an en France. L’incidence augmente régulièrement jusqu’à l’âge de 20 ans en raison de l’identification tardive des formes modérées. Le sex-ratio est en défaveur des garçons (60 % de garçons pour 40 % de filles), sauf dans les cas de retard mental profond où on retrouve le même pourcentage. Cette différence s’explique par les formes de retard mental lié au chromosome X. À la recherche d’une étiologie On estime actuellement pouvoir iden-tifier la cause du retard psychomoteur de l’enfant dans environ 50 % des formes sévères et environ 25 % des formes légères (tableau). On distingue les causes acquises des causes génétiques et, parmi ces dernières, les causes chromosomiques, monogéniques ou liées à l’empreinte génomique parentale. Il existe toutefois des discordances dans la littérature quant à la fréquence de répartition des différentes étiologies, liées à de nombreux biais de sélection des séries publiées, comme par exemple, l’absence de couverture de l’ensemble de la population, et à l’évolution des techniques et des connaissances qui modifient l’impact de certaines étiologies. Les étiologies acquises Les causes acquises de retard psychomoteur sont en régression, comme certaines causes anténatales, en raison des mesures de dépistage et prévention (phénylcétonurie maternelle, infection virale, telle la rubéole, ou parasitaire, telle la toxoplasmose). D’autres causes périnatales anoxo-ischémiques ont régressé en raison des progrès de la réanimation néonatale. Devant une notion de souffrance périnatale avec score d’Apgar bas, il convient d’avoir à l’esprit la notion que les enfants porteurs d’anomalies du développement embryonnaire d’origine génétique ont souvent une mauvaise adaptation à la vie extra-utérine, bien que le retard mental soit inclus dans le déterminisme du syndrome. Il persiste des causes anténatales classiques comme le syndrome d’alcoolisme fœtal ou des prises de médicaments durant la grossesse comme certains anticomitiaux (ex : valproate de sodium, hydantoïnes, etc.). On voit également apparaître de nouveaux agents tératogènes médicamenteux comme par exemple le misoprostol(2) ; il peut en outre exister une suspicion vis-à-vis de produits utilisés dans certains milieux professionnels comme par exemple des agents volatils type éthers de glycols. Les causes postnatales se partagent entre les causes traumatiques, infectieuses, toxiques, tumorales, anoxo-ischémiques et « sociales ». Les enfants porteurs d’anomalies du développement embryonnaire d’origine génétique ont souvent une mauvaise adaptation à la vie extra-utérine. Étiologies génétiques Anomalies chromosomiques Les remaniements chromosomiques identifiables sur le caryotype sont facilement détectables. La plupart des séries estiment que 10 à 15 % des cas de retard mental sont liés à une anomalie chromosomique identifiable sur un caryotype standard. Les anomalies de nombre des chromosomes (aneuploïdies) incluent essentiellement des trisomies et des monosomies partielles. Les plus fréquentes sont les trisomies 21, 18 et 13 et les monosomies 4p (syndrome de Wolf-Hirschhorn) et 5p (maladie du cri du chat). L’incidence de la trisomie 21, classiquement de l’ordre de 1/750 naissances, a chuté de moitié en raison de la politique de dépistage anténatale par le dosage des marqueurs sériques dans le sérum maternel et la mesure de la clarté nucale au premier trimestre de la grossesse. L’incidence de la trisomie 21 a chuté de moitié depuis le dépistage anténatal biologique et échographique. Des micro-remaniements chromosomiques sont de plus en plus identifiés, permettant de caractériser de nouveaux phénotypes microdélétionnels. Certains sont identifiables sur l’examen clinique et le phénotype comportemental par la technique d’hybridation in situ en fluo-rescence (FISH). On peut citer le syndrome de Williams associant dysmorphie faciale, sténose aortique supravalvulaire et phénotype comportemental particulier avec logorrhée et inhibition de la peur de l’étranger, ou encore le syndrome de Smith-Magenis avec troubles du sommeil et troubles du comportement à type d’auto-mutilations(3). Anomalies de l’empreinte génomique parentale Certaines maladies sont liées à un phénomène de régulation d’expression des gènes qui ne s’expriment pas de la même façon sur le chromosome paternel et sur le chromosome maternel. On peut citer par exemple : – le syndrome de Prader-Willi associant hypotonie néonatale, retard psychomoteur modéré, hypogénitalisme et évoluant avec un risque d’obésité ; – et le syndrome d’Angelman avec retard mental sévère, ataxie et rires inappropriés. Ces deux syndromes sont liés à une anomalie de l’empreinte parentale de la même région chromosomique 15q11-q12. Génopathies • Tous les modes de transmission héréditaire mendélienne peuvent se rencontrer. Parmi les maladies à transmission dominante, citons la dystrophie myotonique de Steinert ou la sclérose tubéreuse de Bourneville. Dans le groupe des maladies à transmission récessive, les maladies héréditaires du métabolisme doivent être recherchées, d’autant plus qu’une prise en charge thérapeutique est parfois possible. On l’évoquera d’autant plus en cas d’évolutivité, de phase de latence avec régression ou de consanguinité parentale. • Les maladies mitochondriales sont soit dues à une mutation de l’ADN mitochondrial avec une hérédité alors purement maternelle, soit liées à une anomalie d’un gène mitochondrial nucléaire avec une hérédité récessive autosomique. • Les retards mentaux liés au chromosome X (RMLX) sont caractérisés par une hérédité récessive ou dominante liée au sexe, avec une expression clinique chez le garçon et une transmission par les femmes conductrices, ce qui est le plus souvent identifiable sur la construction d’un arbre généalogique. L’incidence des RMLX serait de l’ordre de 1/1 000 naissances. Les RMLX sont un ensemble de pathologies cliniquement différentes liées à des mutations dans des gènes différents. Certains RMLX sont totalement isolés, sans autre signe détectable, et sont appelés non spécifiques. D’autres s’associent à des variations morphologiques mineures (dysmorphies), ou plus importantes (malformations) qui peuvent orienter le diagnostic étiologique et correspondent aux formes syndromiques des RMLX. On connaît actuellement plus de 200 formes identifiées de RMLX et plusieurs dizaines de gènes sont identifiés sur le chromosome X dans le déterminisme de ces pathologies(4). Le plus fréquent reste le syndrome de l’X fragile qui touche un garçon sur 4 000 et une fille sur 7 000 et qui se caractérise par une triade clinique associant dysmorphie faciale modérée, retard mental avec retard de langage et hyperactivité, et macro-orchidie post-pubertaire. La cause en est une mutation instable de l’ADN sous forme d’un expansion trinucléotidique (CGG) dans la région non codante du premier exon du gène FMR1 situé en Xq27.3. Stratégies d’exploration médicale L’approche clinique L’identification d’un enfant présentant un retard psychomoteur par le médecin de famille, les structures de PMI, l’environnement scolaire ou familial conduisent à une évaluation faite par le pédiatre, le neuropédiatre, le pédopsychiatre et/ou le généticien. Il n’existe pas actuellement de réel consensus sur les stratégies les plus efficaces ou les plus raisonnables pour aboutir à un diagnostic étiologique. Il conviendra toujours de débuter par une approche clinique avec construction d’un arbre généa-logique, recueil des antécédents médicaux familiaux et personnels, examen complet de l’enfant avec évaluation neurologique et analyse de la morphologie, notamment crânio-faciale et des extrémités, et approche du phénotype comportemental. Une évaluation neurocognitive et comportementale est toujours souhaitable dans ce cadre. Temps exploratoire Pour la prescription d’investigations complémentaires, plusieurs algorithmes ont été proposés ces dernières années, fondées sur des métaanalyses de la littérature tenant compte des critères de rentabilité des examens proposés et de coût d’investigation(5,6). Il demeure de nombreux désaccords entre ces différentes stratégies, même si un certain consensus peut en être retiré. Les divergences sont essentiellement liées au moment où les propositions sont faites et à l’émergence de techniques nouvelles, mais aussi au pays où se déroule l’étude en raison de variations géographiques de l’épidémiologie, des étiologies, de la disponibilité et du coût des investigations. Les explorations génétiques Les examens qui apparaissent indispensables et qui ont un « haut rendement » sont : – l’étude du caryotype sur lymphocytes ; – la recherche de microdélétion chromosomique, ciblée sur signes d’appel phénotypique, par technique de FISH. La recherche de micro-remaniements subtélomériques possibles par différentes techniques (multi-FISH, MLPA, CGH-array, etc. cf. infra) devraient se généraliser. De même, une recherche de micro-remaniements sur l’ensemble du génome (pangénomique) est certainement une approche à développer dans les années proches par technique de « puces à ADN » (CGH-array). Parmi les études de génétique moléculaire, la recherche d’un syndrome de l’X fragile sera volontiers demandée systématiquement en l’absence de diagnostic et de microcéphalie chez le proband. Quelques situations particulières peuvent être évoquées. Devant l’association à une épilepsie, un syndrome d’Angelman peut parfois être évoqué et une recherche de mutation dans le gène MECP2, responsable du syndrome de Rett chez la fille, peut se discuter. Parmi les gènes de RMLX récemment caractérisés, l’analyse du gène ARX peut être demandée, notamment en cas d’association à une agénésie du corps calleux, une dystonie ou un syndrome de West chez le garçon(7). Les examens à visée morphologique à la recherche d’anomalies associées garde tout son intérêt, notamment en cas de forme syndromique de retard psychomoteur. Dans ce cadre, on proposera : – une échographie abdomino-rénale ; – un examen ophtalmologique avec étude du fond d’œil ; – une imagerie cérébrale (IRM) ; – et une échocardiographie orientée par l’examen clinique ou le diagnostic suspecté. Les examens radiologiques squelettiques sont envisagés selon le contexte clinique (petite taille, suspicion de chondrodysplasie). Un examen neuro-fonctionnel type EEG est volontiers préconisé. Les examens génétiques sont demandés en fonction de leur « rendement » et du contexte clinique et familial. Les examens métaboliques Ils ne sont pas systématiques et dépendent souvent aussi du contexte clinique. En effet, on estime que les investigations à visée métabolique ne sont positives que dans moins de 1 % des cas en l’absence de contexte évocateur. On pourra demander en fonction du contexte : – une chromatographie des acides aminés (CAA) sanguins et urinaires ; – une chromatographie des acides organiques (CAO) urinaires ; – un dosage d’oligo- et de mucopolysaccharides urinaires ; – un dosage des activités enzymatiques lysosomales ; – des investigations péroxysomales (dosage des acides gras à très longue chaîne, de l’acide pipécolique, de l’acide phytanique, etc.) ; – une recherche de maladie mitochondriale par dosage des rapports d’oxydo réduction, voire l’étude de la chaîne respiratoire dans la biopsie musculaire. De nouvelles maladies métaboliques ont été récemment identifiées et peuvent être intéressantes à rechercher, comme une anomalie de synthèse des précurseurs du cholestérol, dont le chef de file est le syndrome de Smith-Lemli-Opitz, ou encore, une anomalie de glycosylation des protéines sériques (N- et O-glycosylation). De même, un déficit en créatine peut être recherché. « Nouveaux » syndromes microdélétionnels Les nouvelles techniques de résolution chromosomique et moléculaire ont permis de caractériser des aneusomies segmentaires assez méconnues jusqu’à maintenant. Ces techniques incluent la FISH (figure 1), l’hybridation génomique comparative sur puces à ADN (microarray-CGH) (figure 2) et des techniques de génétique moléculaire comme la QM-PSF (Quantitative Multiplex PCR of Short Fluorescent Fragments). Grâce à ces outils, de nouveaux syndromes cliniques microdélétionnels des régions chromosomiques subtélomériques ont pu être identifiés. Trois syndromes microdélétionnels s’avèrent assez communs : les délétions 1p36, 2q37 et 22q13. Figure 1. Identification d’une microdélétion 1p36 par FISH (photo L. Taine). Figure 2. Identification d’une microdélétion du gène CREBBP par la technique de CGH-array (photo B. Arveiler). Monosomie 1p36 La monosomie 1p36 est probablement un syndrome des gènes continus et son incidence de novo est de 1/5 000 à 1/10 000 nouveau-nés. Le phénotype neurologique est au devant de la scène avec un retard psychomoteur le plus souvent sévère, une hypotonie, une épilepsie quasi constante (crises myocloniques, tonico-cloniques généralisées, focales hémicorporelles, partielles complexes, spasmes infantiles) et des troubles du comportement à type d’auto-agressivité(8). Le retard de croissance est le plus souvent post-natal et l’évolution associe microcéphalie et tendance à la surcharge pondérale. La dysmorphie faciale est l’élément qui permet d’évoquer le diagnostic, associant brachycéphalie, sourcils horizontaux, yeux enfoncés dans les orbites, fentes palpébrales courtes, ensellure nasale marquée, hypoplasie de l’étage moyen de la face, menton pointu, coins des lèvres tombants, oreilles dysplasiques (figure 3). D’autres signes peuvent être associés, comme une cardiomyopathie ou une autre anomalie cardiaque, une surdité, une brachydactylie, une hypothyroïdie ou plus rarement une fente labio-palatine (10 à 40 % des cas). La microdélétion du chromosome 1p36 correspond généralement à une haplo-insuffisance d’une région de 0,5 mégabase (Mb). Figure 3. Aspect facial de face d’un enfant atteint de monosomie 1p36. Noter les sourcils horizontaux et les yeux enfoncés dans les orbites. Monosomie 2q37 La monosomie 2q37 est aussi connue sous le nom de syndrome ODA-like (ostéodystrophie héréditaire d’Albright) en raison de la brachydactylie caractéristique associée. Cette brachymétacarpie des 3e, 4e et 5e rayons est cependant inconstante. L’hypotonie et le retard mental sont les manifestations les plus fréquentes du syndrome(9). Des troubles du comportement (hyperactivité, stéréotypies, agressivité) sont volontiers présents, pouvant mimer un syndrome autistique(10). Les autres signes associés sont : obésité, petite taille, dysmorphie faciale avec visage lunaire et sourcils marqués dans la partie proximale, et parfois anomalies cardiaques et génitales. La région minimale critique de délétion est de l’ordre de 1 Mb. Microdélétion 22q13.3 La microdélétion 22qter a initialement été identifiée par la cytogénétique constitutionnelle. Elle concerne typiquement la bande terminale 22q13.3 et a été associée à des translocations familiales et de novo. La FISH a permis de diagnostiquer des patients avec des phénotypes compatibles, non détectés par la cytogénétique conventionnelle. Le syndrome de délétion 22q13 associe principalement un retard global du développement mental, une hypotonie généralisée, un langage absent ou très réduit et une croissance normale ou en avance(11). Les anomalies mineures incluent une dolichocéphalie, un ptosis, des oreilles dysplasiques, une anomalie de croissance des ongles, un menton pointu, une syndactylie 2-3 des orteils et de grandes mains. Une tolérance accrue à la douleur est notée et des convulsions sont retrouvées chez 30 % des patients. Un syndrome autistique est fréquemment associé et peut être le signe révélateur de la maladie(12). Le chromosome 22 en anneau peut présenter le même phénotype que le syndrome de délétion 22qter, mais l’expressivité est compliquée par le mosaïcisme souvent associé au chromosome en anneau. La taille de la délétion varie de 130 Kb à plus de 9 Mb et le gène PROSAP2 (SHANK3) est inclus dans la région critique. Ce gène codant pour une protéine de structure post-synaptique, pourrait être impliqué dans le phénotype neurologique, et notamment dans l’absence de développement du langage(13). Les nouvelles techniques d’étude du génome permettent de plus en plus d’identifier un déterminisme moléculaire à des syndromes cliniquement répertoriés, ou peuvent caractériser un micro-remaniement permettant de décrire un nouveau phénotype clinique. Cela est vrai actuellement pour les microdélétions des régions subtélomériques, et se développe au niveau pangénomique. D’autres syndromes microdélétionnels ou aneusomies segmentaires voient le jour, comme la délétion 9q34 par exemple.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :





