Publié le 09 avr 2015Lecture 11 min
Troubles du rythme dans la famille : que faire pour la descendance ?
V. LUCET, C. DENIS, H. NGAYAP, Unité de rythmologie pédiatrique, Centre pédiatrique des Côtes, Les Loges-en-Josas
Depuis 20 ans, les progrès de la biologie moléculaire ont permis une meilleure compréhension et une meilleure classification des troubles du rythme cardiaques héréditaires. Liées le plus souvent à des anomalies de canaux potassiques, sodiques ou calciques, ces canalopathies sont responsables de troubles du rythme rares, mais graves, pouvant mettre en jeu le pronostic vital. La prise en charge familiale doit donc se faire, idéalement, dans des centres spécialisés, disposant d’une équipe multidisciplinaire et d’une expertise.
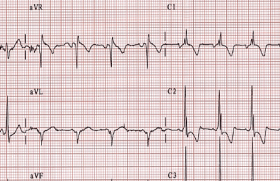
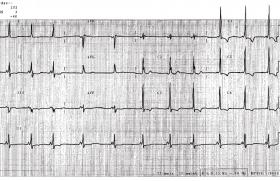
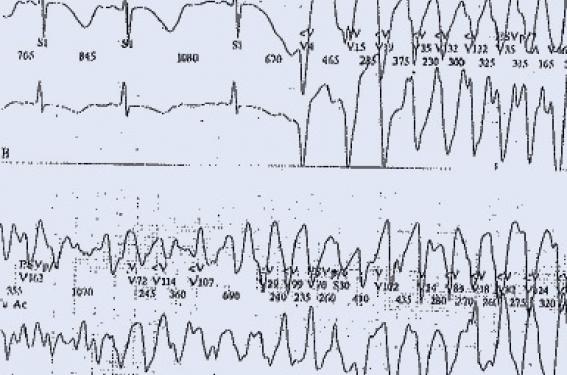
Principaux troubles du rythme héréditaires Syndrome de QT long Le Syndrome de QT long associe une prolongation de l’espace QT (QTc ≥ 440 ms), un risque de torsade de pointe et de mort subite familiale (figure 1). La mesure de l’espace QT doit donc faire partie de l’analyse systématique de l’électrocardiogramme de l’enfant : Un QTc ≥ 450 ms chez l’enfant doit être considéré comme pathologique en l’absence d’autres causes susceptibles d’allonger le QT. La fréquence de ce syndrome est estimée actuellement à 1/2 500 individus. Au moins 13 gènes ont été mis en cause depuis 1995, mais trois d’entre eux constituent 75 à 80 % des mutations documentées. Le plus fréquent est le LTQ 1 (35 % des cas). Il correspond à une perte de fonction du canal potassique. Figure 1. QT long et torsade de pointe. Il est responsable de syncopes déclenchées par le stress ou l’effort. Le LQT2 (25 à 30 % des cas) donne classiquement des syncopes déclenchées par l’émotion ou les stimuli auditifs. Il est lié à une perte de fonction du canal potassique, alors que le LQT3 moins fréquent (5 à 10 % des cas) est en rapport avec un gain de fonction du canal sodique et peut être responsable de morts subites nocturnes. Les 10 autres syndromes de QT long sont beaucoup plus exceptionnels et sont responsables d’environ 5 % des mutations identifiées. Parmi eux, il faut citer le syndrome de JervellLange Nielsen (autosomique récessif avec surdité) retrouvé dans un 1 cas/million, et responsable de formes sévères de QT long, le syndrome de Andersen avec extrasystoles ventriculaires polymorphes d’effort (LQT7) et les syndromes de Timothy (LQT8) avec syndactylie. Les tachycardies ventriculaires polymorphes catécholergiques Caractérisées par la survenue de syncope ou de mort subite à l’effort ou à l’émotion, les tachycardies ventriculaires polymorphes catécholergiques (TVPC) sont typiquement des arythmies du grand enfant (> 6 ans) à l’examen clinique et échographique normaux. L’électrocardiogramme de repos est lui aussi normal, mais l’effort déclenche de façon répétitive des extrasystoles polymorphes, typiquement bidirectionnelles, pouvant dégénérer en fibrillation ventriculaire (figure 2). Dans 65 % des cas, on retrouve chez le cas-index une mutation de RyR2 affectant la perméabilité du canal calcique membranaire. Cette forme est à transmission dominante. Les formes autosomiques récessives (CAS Q2) sont beaucoup plus rares (5 % des cas). Figure 2. Tachycardie ventriculaire polymorphe. Les autres canalopathies rythmiques Elles sont beaucoup plus rarement observées chez l’enfant. Le syndrome de Brugada Caractérisé par un sus-décalage particulier de ST en précordiale droite (figure 3) et un risque de syncope ou de mort subite familiale, le syndrome de Brugada est rare chez l’enfant. Sa fréquence est estimée à 1/5 000 individus environ. Plusieurs formes électriques ont été décrites, mais seul le type 1 (spontané ou révélé par un test à la Flécaine®) est caractéristique. Les mutations documentées ont été retrouvées sur au moins 8 gènes, mais les plus fréquentes (75 % des cas connus) concernent le gène SCN5A. Figure 3. Syndrome de Brugada. Les troubles conductifs progressifs familiaux D’apparition progressive, les troubles conductifs progressifs familiaux peuvent être soit isolés, soit associés à une atteinte cardiaque (communication inter-auriculaire, myocardiopathie, myopathie, laminopathie). La recherche d’une mutation peut être discutée quand le caractère familial des troubles conductifs est établi ou qu’il existe une pathologie associée évocatrice. Le syndrome du QT court Très rare, ce syndrome est de description récente (2000). Il est caractérisé par un QT très court (figure 4), sans retour à la ligne isoélectrique du segment ST, avec des ondes T amples et pointues. Il s’y associe un risque de fibrillation auriculaire et de mort subite familiale, parfois précoce, chez le nourrisson. Actuellement, trois gènes ont été identifiés, responsables d’une anomalie du canal potassique à transmission autosomique dominante, mais d’expressivité variable. Figure 4. Syndrome de QT court. La dysplasie ventriculaire droite arythmogène Malgré les progrès réalisés ces dernières années, la dysplasie ventriculaire droite arythmogène (DVDA) reste un ensemble flou, tant sur le plan clinique que génétique. D’apparition progressive au cours de la vie et d’expressivité variable, la DVDA est caractérisée par l’existence d’anomalies de l’électrocardiogramme (onde epsilone, l’inversion des ondes T en précordiale droite), le risque de survenue d’arythmie ventriculaire grave favorisée par l’effort et l’apparition progressive d’altérations du ventricule droit (inflammation, amincissement des parois, infiltration fibro-adipeuse, dilatation, anévrysmes, hypertrophie des trabéculations, etc.), documentées par les techniques d’imagerie (IRM, angiographie) ou l’autopsie. La plupart des gènes impliqués codent pour les protéines desmosomales (70 %), mais une grande variété de mutations à transmission dominante ont été décrites, faisant parfois le lien avec des formes familiales de myocardiopathies ou les TVPC. Nécessité et limites d’une enquête familiale Si l’un des syndromes cités plus haut est découvert ou suspecté, il apparaît important d’effectuer une enquête familiale avec la réalisation d’un examen clinique, d’un électrocardiogramme et éventuellement d’une échographie aux apparentés du premier degré. Cela permet parfois de confirmer le diagnostic en cas de découverte d’autres cas semblables et d’organiser une prise en charge thérapeutique adaptée. Pour autant, un bilan cardiologique initial normal ne permet pas d’éliminer le diagnostic : on estime, par exemple, que 10 à 40 % des sujets atteints d’un syndrome de QT long, on un QTc dans les limites de la normale. Il en est de même pour les sujets atteints de TVPC qui peuvent avoir un ECG d’effort normal ou ne présenter que quelques rares extrasystoles ventriculaires isolées, ce qui ne permet pas d’éliminer la possibilité d’une syncope ou d’une mort subite parfois précoce, confirmée par l’autopsie moléculaire dans certains cas de mort subite du nourrissons (MSN) : 15 % des MSN avec autopsie négative seraient dues à des canalopathies héréditaires : QT long, TVPC, etc. L’aspect typique sur l’électrocardiogramme du syndrome de Brugada peut n’être qu’intermittent et démasqué par les épisodes fébriles. La dysplasie ventriculaire droite peut être parfaitement silencieuse pendant des décennies. Il en est de même pour les troubles conductifs progressifs familiaux. C’est dire l’intérêt de compléter l’enquête clinique par une enquête moléculaire. Nécessité et limites de l’enquête moléculaire Si une mutation pathogène a été identifiée chez le cas-index, la recherche d’autres sujets atteints dans la famille ne se discute pas, quel que soit l’âge dans le syndrome du QT long, le syndrome du QT court et les TVPC. Probablement sans urgence dans les autres canalopathies, rarement symptomatique dans l’enfance. En cas de découverte fortuite d’un QT long isolé et en l’absence de symptôme ou d’antécédents familiaux, on prescrit une recherche moléculaire (limitée dans un premier temps aux trois formes les plus fréquentes de QT long) si le QTc est ≥ 480 ms chez l’enfant et 500 ms chez l’adulte. Ces mêmes recherches peuvent être discutées pour des QTc moins prolongés (QTc > 460 ms chez l’enfant). L’absence de mutation retrouvée chez le cas-index n’élimine cependant pas le diagnostic. Dans le syndrome du QT long, la « rentabilité » des tests génétiques n’est que de 80 %. Elle est d’environ 60 à 70 % dans les TVPC typiques et les DVDA. Elle tombe à 20 % dans le syndrome de Brugada. Or, ces recherches sont compliquées et chronophages, compte tenu notamment de la grandeur des gènes à explorer (SCN5A, RyR2) et du grand nombre de mutations possibles (plus de 100 mutations identifiées sur le gène RyR2 des TVPC). De plus, il faut savoir qu’il existe un nombre non négligeable de mutations non pathogènes ou de significations incertaines dans la population témoin (entre 2 et 4 % dans les QT long et les TVPC, jusqu’à 16 % de faux positifs dans la DVDA, (tableau). Nécessité du suivi familial Compte tenu des aléas et des incertitudes qui persistent après une évaluation clinique de la famille d’un sujet atteint, il apparaît important de mettre en place un suivi régulier non seulement du « cas témoin » et des autres patients dépistés par l’enquête familiale, mais aussi de la fratrie. Pour les parents, la normalité de l’examen clinique, de l’électrocardiogramme et l’échographie est un élément rassurant en l’absence de signes fonctionnels ou d’antécédents familiaux chez leurs propres apparentés (parents, fratrie). Pour les frères et sœurs d’un enfant porteur d’une canalopathie rythmique, on peut conseiller la mise en place d’un suivi cardiologique non anxiogène au long court, peu contraignant pendant l’enfance, avec, par exemple, un bilan tous les 2 ans. Ce n’est souvent qu’à l’adolescence que l’on propose, dans un deuxième temps, des examens plus approfondis (test de provocation à la Flécaine® pour les syndromes de Brugada, IRM ou angiographie en cas de suspicion de DVDA). Nécessité du traitement Compte tenu de la possibilité de révélation très précoce d’un syndrome du QT long ou d’une TVPC, par un trouble du rythme ventriculaire grave ou une mort subite du nourrisson, il est important de débuter un traitement préventif par bêtabloquant le plus tôt possible après le dépistage de ces deux canalopathies. L’efficacité d’un traitement par bêtabloquant à bonne dose, régulièrement réévalué, est prouvée dans le syndrome du QT long (essentiellement LQT1 et LQT2) et les TVPC. Pour le LQT3, l’efficacité préventive des bêtabloquants est moins établie, justifiant parfois un renforcement de la thérapeutique par la Flécaine®. Chez tous les sujets atteints, une liste de médicaments contre-indiqués (disponible sur internet) doit être remise à la famille. En cas de récidive de syncope sous traitement et en cas de TVPC mal contrôlée chez l’adolescent, la mise en place d’un défibrillateur implantable se discute. Les formes symptomatiques non traitées ont une mortalité globale de 50 % à 10 ans et sont d’autant plus sévères que le QTc est long ou qu’il s’agit d’une forme clinique particulière (syndrome de Jervell, forme néonatale avec bloc auriculo-ventriculaires, certains LQT3, etc.). Les autres canalopathies rythmiques asymptomatiques ne nécessitent pas, habituellement, de traitement urgent dans la petite enfance, mais le défibrillateur implantable est parfois la seule solution pour prévenir la mort subite dans certains cas pédiatriques exceptionnels. La pratique du sport Les arythmies familiales à composante adrénergique (QT long, TVPC, DVDA) contre-indiquent la pratique du sport et, en particulier de la natation, compte-tenu d’un taux élevé de noyades rapportées dans la littérature. Il importe, cependant, pour l’équilibre psychologique de l’enfant, de le laisser jouer et pratiquer les activités physiques récréatives, notamment avec sa famille. Il n’est pas prouvé, en revanche, que le sport de loisirs soit particulièrement dangereux dans le syndrome de Brugada. La grossesse L’immense majorité des canalopathies rythmiques familiales concernent des sujets sains au développement physique et intellectuel normal. Elles sont d’autre part accessibles à des thérapeutiques relativement simples. Pour ces raisons, sauf cas particulier, le dépistage in utero et l’interruption médicale de grossesse ne sont pas recommandées (de même que le diagnostic préimplantatoire). Toutefois, les futures mamans porteuses d’une mutation reconnue doivent pouvoir bénéficier d’un suivi spécialisé de leur grossesse et accoucher dans des maternités susceptibles de prendre en charge un trouble du rythme néonatal ou les complications éventuelles d’un traitement bêtabloquant chez la maman. Conclusion Sous condition d’une bonne prise en charge par des équipes spécialisées, les enfants atteints de canalopathies rythmiques héréditaires doivent pouvoir mener une vie strictement normale. Les contraintes de la surveillance cardiologique et du traitement médical au long court sont réelles, mais surmontables. L’indication d’un soutien psychologique temporaire peut se discuter, dans ces familles, mais est rarement nécessaire.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :