Publié le 16 sep 2012Lecture 9 min
Une énurésie… secondaire
D. ARMENGAUD, CHI de Poissy/Saint-Germainen- Laye

Lounes est un garçon de 5 ans, adressé par son médecin traitant qui, s’inquiétant d’une prise pondérale insuffisante, dans un contexte de polyurie avec polydipsie et reprise d’une énurésie intermittente, a fait pratiqué un bilan biologique. Celui-ci retrouve un ionogramme normal avec une urée à 14,1 mmol/l, une créatininémie à 132 μmol/l…
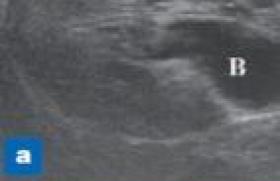
Histoire clinique Lounes est né à 41 semaines d’aménorrhée au poids de 3 180 g pour une taille de 51 cm. Il a été hospitalisé à 16 mois pour une laryngite aiguë non compliquée et a présenté quelques épisodes de dyspnée expiratoire répondant à un traitement bronchodilatateur, administré par chambre d’inhalation sans avoir eu besoin de recourir à une corticothérapie inhalée au long cours. À l’interrogatoire, il semble bien que cet enfant ait toujours pris des boissons abondantes, mais depuis 4 à 5 mois cela a pris plus d’ampleur, avec notamment une nécessité de boire la nuit qui n’existait pas auparavant. À l’examen, on est face à un enfant en bon état général, ne se plaignant d’aucun trouble fonctionnel. Le poids est de 16,2 kg (- 1 DS) pour une taille de 106,5 cm (- 0,5 DS). L’examen somatique est sans particularité. Il n’y a pas d’amaigrissement récent (cf. courbe de croissance reconstituée [figure 1]), ni signes de dénutrition (IMC à 14,3), ni de déshydratation patente. L’auscultation cardio-pulmonaire est normale, la pression artérielle à 114/ 64 mmHg. L’examen neurologique est normal. Une bandelette urinaire, réalisée compte tenu de la notion de polyurie, ne retrouve pas de glycosurie. Un bilan biologique est prélevé. Examens biologiques NFS : – Hb : 9,4 g/100 ml ; – GR : 34 800 000/mm3 ; VGM 79 μ3 ; – GB : 6 400/mm3 (61 % de PNN) ; – Plaquettes : 322 000/mm3 Glycémie : 4,7 mmol/l Ionogramme sanguin (mmol/l) : Na : 143 ; K : 4,3 ; bicar : 18 ; urée : 16,8 ; calcémie : 2,10 ; phosphorémie : 2,52 Protides : 70 g/l Créatinine : 144 μmol/l BU : protéinurie : traces ; hématurie négative CRP : 20 mg/l Examens d’imagerie Écho rénale Figure 1. Courbe de croissance. Quelles hypothèses étiologiques peuvent-elles être formulées ? Hypothèses diagnostiques Il existe manifestement une insuffisance rénale qui est parfaitement tolérée, ne donnant aucun des symptômes inquiétants d’insuffisance rénale aiguë, avec notamment absence : – de syndrome oedémateux ou de prise de poids récente ; – d’hypertension artérielle, dont le contrôle des chiffres en continu restera normal ; – d’oligo-anurie, avec au contraire, la notion d’une polyurie depuis quelques mois sans retentissement clinique évident en dehors de la reprise d’une énurésie qui semble donc bien « secondaire » ; La reconstitution de la courbe de croissance (figure 1, p. 6) ne montre pas de ralentissement de la croissance staturale, mais un certain degré de maigreur puisque l’IMC à 14,3 se situe aux alentours du 10e percentile. Pour essayer d’avancer sur le plan étiologique, la réflexion doit se fonder sur le « maître symptôme », qui n’est pas une énurésie, mais un syndrome polyurodipsique, qui doit faire évoquer en premier lieu : une polyurie osmotique et donc un diabète sucré, a priori insulinoprive à cet âge, mais dont la révélation parfois progressive sur quelques jours ou semaines, se fait rarement sur quelques mois. Elle s’accompagne en outre de signes cardinaux, que sont l’amaigrissement, la déshydratation, l’altération de l’état général et biologiquement, une cétose et/ou une acidose avec glycosurie secondaire à l’hyperglycémie, tous signes absents ici. Beaucoup plus rarement, une polyurie liée à une hypercalcémie (généralement d’origine maligne) ou une hypokaliémie, toutes deux également absentes du bilan biologique pratiqué. Une polyurie insipide qui se caractérise par une osmolarité urinaire basse, inadaptée, car contemporaine d’une élévation de l’osmolarité plasmatique. La recherche de son origine nécessiterait une mise en observation pour réaliser un bilan des entrées/ sorties et une épreuve de restriction hydrique, qui permettrait de différencier : • un diabète insipide d’origine centrale par défaut de synthèse ou de sécrétion de l’hormone antidiurétique (ADH) : – soit « idiopathique », de transmission génétique autosomique dominante (à évoquer et surveiller régulièrement lorsqu’un cas index a été diagnostiqué), débutant entre 1 et 6 ans ; un test de restriction hydrique, réalisé sous haute surveillance, montrerait l’absence de régulation (> natrémie, > osmolarité sanguine, taux de vasopressine indétectable) ; l’IRM montrerait en hyper-signal de la post-hypophyse (accumulation de l’hormone non mature) ; il existerait une sensibilité à l’arginine vasopressine exogène, permettant le traitement « substitutif » ; – soit le défaut de synthèse d’ADH est secondaire : acquis (post-traumatique, postopératoire) ; tumoral en rapport avec un craniopharyngiome (se révélant par un ralentissement de croissance statural et/ou des signes d’hypertension intracrânienne) ; ou dans le cadre d’une histiocytose X, d’une sarcoïdose ou d’une auto-immunité. • Une polydipsie primaire souvent d’installation progressive (contexte psychoaffectif), dont le diagnostic différentiel avec un diabète insipide est parfois difficile : lorsque ce trouble est installé, la potomanie entraîne une inflation du secteur extracellulaire qui induit une inhibition de la sécrétion d’ADH ; l’importance de la diurèse diminue l’efficacité du gradient de concentration cortico-médulaire. • Un diabète insipide néphrogénique (périphérique), par insensibilité des cellules tubulaires à l’ADH, dont la transmission est récessive, autosomique ou liée à l’X. Un accident de déshydratation hypernatrémique survient alors souvent dès les premières semaines de vie, avec une osmolarité urinaire inadaptée restant toujours < 300 mosm. • De nombreuses affections rénales en rapport avec des tubulopathies interstitielles chroniques, accompagnées ou révélées par une polyurie inadaptée entraînant une polydipsie réactionnelle. S’il existe une insuffisance rénale, elle est le plus souvent de type prérénale, sans altération de la fonction glomérulaire, tout du moins au début… : uropathies malformatives obstructives, retentissant sur le tubule distal ; tubulopathies, syndrome de Toni-Debré- Fanconi (cystinose) ; IR d’origine médicamenteuse, concernant rarement l’enfant (lithium…). Une maladie rénale, la néphronophtise, par élimination successive de ces diagnostics, peut être évoquée sur l’histoire de Lounes. Le début retardé dans la forme juvénile la plus fréquente, l’absence de poussée hypertensive, d’hématurie, de syndrome oedémateux en font une maladie spécialement silencieuse en dehors d’une polyurie « facile à compenser »… Le bilan biologique confirme l’existence d’une insuffisance rénale « organique » avec une clairance calculée de la créatinine à 35 ml/min/1,73 m2 et une hyperphosphatémie (défaut d’élimination tubulaire). Figure 2. Échographie rénale : rein de taille normale, avec hyperéchogénicité et dédifférenciation cortico-médullaire. A : rein droit (hyperéchogénicité par rapport au foie). B : rein droit en coupe transversale. C : rein gauche de taille normale. L’échographie rénale (figure 2) montre un aspect dédifférencié du parenchyme rénal, qui non pathognomonique, est très en faveur d’une pathologie tubulointerstielle. Commentaire La néphronophtise est une néphropathie tubulo-interstitielle chronique d’origine génétique se transmettant sur le mode autosomique récessif. Sa prévalence est rare (1/100 000), mais son évolution vers l’insuffisance rénale terminale avant l’âge de 15 ans fait qu’elle représente près de 15 % des transplantations rénales pédiatriques. La forme juvénile, qui est la plus fréquente, débutant après 2 ans (ce qui la différencie de la forme infantile), se caractérise par des manifestations cliniques très pauvres en dehors d’un trouble de concentration des urines progressif. Ce dernier passe souvent inaperçu (il a l’habitude de boire beaucoup) ou est une « cause » d’énurésie… qui impose toujours une démarche diagnostique d’élimination rigoureuse. Parfois, c’est au cours d’une gastro- entérite avec déshydratation sévère et inhabituellement hyponatrémique, que la maladie se révèle avec une altération de la fonction rénale, qui n’est pas seulement « fonctionnelle ». Ce peut être aussi par un retard de croissance statural, voire pubertaire, que le diagnostic peut être porté, mais l’absence d’hypertension artérielle, de protéinurie ou d’hématurie sont des silences qui ne font pas en règle évoquer une maladie rénale. Les progrès de la génétique ont permis d’identifier plusieurs gènes délétés, dont le premier identifié (NPHP1) localisé en 2q13 est présent dans 70 % des cas, ce qui permet souvent de ne plus avoir besoin de recourir à la biopsie rénale pour le diagnostic. D’autres anomalies géniques ont été identifiés, comme NPHP1-9, dont la présence dans d’autres tissus explique pour une part des associations cliniques décrites (dégénérescence tapétorétinienne avec rétinite pigmentaire, atrophie cérébelleuse…). Les fonctions de ce gène sont en cours de découverte, avec notamment un rôle dans la synthèse de protéines de jonction intercellulaire, mais aussi dans le fonctionnement du cil primaire des cellules tubulaires, donnant une base génétique commune à toutes les maladies rénales s’accompagnant de la formation de kystes. Évolution Chez Lounes, le diagnostic de néphronophtise sera confirmé par la mise en évidence par PCR d’une délétion homozygote du gène NPHP1. L’évolution de l’insuffisance rénale progressive et inexorable vers l’insuffisance rénale terminale, malgré la prise en charge en milieu spécialisé et l’instauration de mesures de protection rénale, conduira à une transplantation rénale à l’âge de 9 ans, dont on peut espérer le bon pronostic à long terme en l’absence de risque de récidive sur le greffon.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :