Allergologie - Immunologie
Publié le 18 oct 2019Lecture 8 min
Déficits immunitaires constitutionnels et allergies alimentaires
Étienne MERLIN, Pédiatrie générale multidisciplinaire, CHU Clermont-Ferrand
Les anomalies génétiques du système immunitaire, ou déficits immunitaires constitutionnels (DIC), regroupent plus de 300 maladies et plus de 5 000 patients en France. Ces maladies génétiques n’exposent pas seulement à un risque infectieux, mais également à des maladies auto-immunes, inflammatoires et allergiques. L’allergie alimentaire est très fréquente au cours du déficit lié à une mutation de DOCK8, dont le phénotype associe dermatite inflammatoire sévère, infections virales notamment herpétiques, et hyper-IgE. Une entéropathie compatible avec une allergie alimentaire sévère chez le nouveau-né ou le nourrisson, en particulier de sexe masculin, doit faire évoquer un syndrome de Wiskott-Aldrich (eczéma, thrombopénie à microplaquettes, otites récidivantes) ou un IPEX (polyendocrinopathie autoimmune liée à l’X) et justifie d’un avis rapide auprès d’un pédiatre immunologiste.
L’allergie alimentaire est une réaction anormale et spécifique de l’organisme au contact d’une substance étrangère qui n’entraîne pas de trouble chez la plupart des sujets. Les réactions aiguës, qui peuvent mener à l’anaphylaxie, impliquent le plus souvent une médiation IgE. Cette implication des IgE n’est en revanche pas toujours présente au cours des réactions tardives (œsophagites et gastro-entérites à éosinophiles). Enfin, le syndrome d’entérocolite induit par les protéines alimentaires (SEIPA) n’implique pas de médiation IgE. Il peut réaliser des formes aiguës (débâcle diarrhéique, vomissements, hypotension et fièvre survenant dans les 2 heures suivant l’ingestion) ou chroniques (vomissements intermittents, diarrhées, irritabilité, mauvaise prise de poids etc.)(1). L’homéostasie immunitaire repose sur un équilibre fragile entre des populations effectrices et des populations régulatrices. Les déficits immunitaires constitutionnels (DIC) n’exposent pas seulement à un risque infectieux, mais également à des désordres immunologiques conduisant à des maladies auto-immunes, inflammatoires, et allergiques dans environ 25 % des cas, et ce sans différence notable selon que le déficit affecte l’immunité innée ou acquise, humorale ou cellulaire(2).
Les maladies génétiques du système immunitaire exposent à des infections, et aussi à des maladies auto-immunes, inflammatoires et des allergies.
Parmi ces manifestations dysimmunitaires, l’allergie alimentaire est peu fréquente, voire moins fréquente que dans la population générale(3). Cependant, le chevauchement entre allergie alimentaire et DIC peut conduire à 3 situations cliniques :
– une authentique allergie alimentaire peut s’intégrer dans un tableau clinique devant faire évoquer le diagnostic de DIC, ou survenir chez un enfant connu comme porteur d’un DIC ;
– certains déficits immunitaires peuvent provoquer des manifestations digestives néonatales ou précoces qui peuvent faire évoquer une allergie alimentaire, notamment aux protéines de lait de vache. Dans certains cas, un régime d’éviction peut d’ailleurs temporairement atténuer les symptômes ;
– une authentique allergie peut être la cause d’un tableau faisant discuter un DIC. Le diagnostic de DIC est alors récusé par la réversibilité des anomalies immunitaires à l’arrêt de l’exposition à l’allergène.
L'allergie alimentaire est fréquente au cours de certains DIC
Les syndromes d’hyper-IgE
On distingue deux types de syndromes d’hyper IgE (HIES) selon le mode de transmission (autosomique dominant ou récessif) et les gènes incriminés. Dans les deux cas, le phénotype est assez proche, dominé par des infections à staphylocoque doré ; en particulier abcès « froids », des infections fongiques invasives, une petite taille et une dermatite néonatale éczématoïde. Le taux d’IgE est très élevé. Un score permet de distinguer cette maladie d’une maladie atopique sévère. Les mutations inhibitrices de STAT3 provoquent une maladie à transmission autosomique dominante (AD-HIES), tandis que les mutations de DOCK8 et TYK2 sont responsables de formes à transmission récessive (AR-HIES). En termes d’allergie, en revanche, il existe une différence fondamentale entre les 2 syndromes : alors que les AR-HIES font d’authentiques allergies alimentaires et pneumallergies, avec des IgE spécifiques élevés et une éviction efficace, les enfants atteints d’AD-HIES ont un défaut de mémoire immunitaire et d’activation mastocytaire qui ne permet pas l’installation durable d’une réaction allergique. Le diagnostic d’allergie alimentaire n’est donc pertinent (et fréquent) que chez les AR-HIES. Les allergies alimentaires (œuf, lait, arachide, etc.) affectent jusqu’à 60 % des enfants atteints d’ARHIES(4) ; elles ne sont pas toujours guéries par l’allogreffe de cellules souches hématopoïétiques, voire peuvent apparaître après. Allergie alimentaire + dermatite précoce + abcès + infections virales sévères + IgE très élevés : penser aux syndromes d’hyper-IgE.
L’IPEX
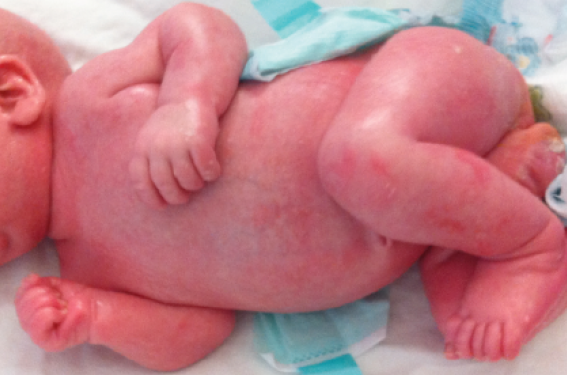
Le syndrome IPEX (polyendocrinopathie immunitaire liée à l’X) est dû à un défaut de lymphocytes T régulateurs par mutation du facteur de transcription FOXP3. Il se révèle dans les mois suivant la naissance par un tableau cutané (éruption éczématiforme, dermatite atopique sévère) et digestif (diarrhée sévère), rapidement complété par une auto-immunité notamment pancréatique et hématologique (anémie, thrombopénie, neutropénie auto-immunes). Si l’atteinte digestive relève classiquement d’un mécanisme auto-immun (attesté par la présence d’anticorps antientérocytes AIE75), d’authentiques allergies alimentaires sont parfois présentes, voire dominent le tableau(5) associant dermatite atopique sévère, réactions anaphylactiques, entéropathie exsudative, élévation des IgE totales et spécifiques, éosinophilie. Le traitement repose sur les immunosuppresseurs, en particulier sirolimus, et l’allogreffe de cellules souches hématopoïétiques. Un phénotype proche de l’IPEX, mais pouvant affecter également les filles, a été rapporté à des mutations d’autres gènes que FOXP3, impliquant un dysfonctionnement des lymphocytes T régulateurs (gènes STAT5b, IL2R, ITCH)(6). Le syndrome de Loeys Dietz Décrit en 2005, ce syndrome est dû à une mutation du gène codant pour une cytokine régulatrice, le TGF bêta(7). Il associe un habitus marfanoïde, un retard des acquisitions modéré, et des manifestations allergiques marquées (en particulier digestives et respiratoires). L’altération de la signalisation du TGF rend compte des manifestations marfanoïdes (de nombreux gènes, notamment du collagène, sont exprimés sous la dépendance de cette signalisation) et du phénotype allergique (dysfonction des lymphocytes régulateurs). Les allergies alimentaires affectent environ 25 % des patients, les allergènes les plus fréquents sont l’œuf, le lait, le soja et l’arachide. Le pronostic de ce syndrome est en grande partie lié à l’atteinte de l’aorte ascendante. Le syndrome de Netherton Ce syndrome de transmission autosomique récessive est dû à la mutation du gène SPINK5, qui code pour un inhibiteur de sérine protéase, exprimé notamment dans les couches superficielles de l’épiderme. Le défaut protéique conduit à une altération importante de la barrière cutanée. Le tableau associe une érythrodermie congénitale (figure 1) à des atteintes infectieuses notamment à staphylocoque, qui relèvent de l’atteinte cutanée, d’une part, et de déficit immunitaire, d’autre part, des diarrhées sont également fréquentes. Le diagnostic repose sur l’examen d’un cheveu « en bambou ». La prévalence de l’allergie alimentaire est élevée. Elle peut se manifester sous la forme d’anaphylaxie, d’œsophagite à éosinophiles, angioœdème et s’associer à une élévation importante des IgE totales(8). Le déficit sélectif en IgA Le déficit sélectif en IgA est fréquent (prévalence estimée d’environ 1/200), et généralement considéré comme asymptomatique. Cependant, une fréquence accrue de laryngites et d’allergies alimentaires (rapportées par les parents) par rapport à une population témoin (33 % versus 12 %) a été rapportée dans une étude suédoise. Cependant, il n’y avait pas d’augmentation des IgE spécifiques, suggérant un mécanisme non-IgE médié. Le rôle des IgA dans la prévention des allergies alimentaires n’est pas certain.
Figure 1. Érythrodermie néonatale au cours d’un syndrome de Netherton.
Entéropathie précoce : ne pas porter trop vite le diagnostic d'allergie alimentaire
L’allergie aux protéines de lait de vache est la cause la plus fréquente d’entéropathie dysimmunitaire non-infectieuse chez un nouveau-né ou un nourrisson. Cependant, une entéropathie néonatale peut révéler un DIC, et ne pas relever d’un mécanisme purement allergique. L’atteinte digestive est susceptible d’être améliorée par l’éviction des protéines du lait de vache, ce qui peut entretenir la confusion diagnostique. Cette confusion entre APLV et DIC peut entrainer un retard diagnostique préjudiciable(9). Chez l’enfant plus grand, les atteintes digestives des déficits immunitaires prennent plus volontiers le masque d’une maladie inflammatoire chronique que d’une maladie allergique. Les maladies monogéniques responsables d’entérocolite néonatale auto-immune non-allergique sont de l’ordre d’une dizaine (tableau 2)(10). Les éléments évocateurs sont une diarrhée glairo-sanglante éventuellement fébrile, une inflammation biologique, une mauvaise croissance staturo-pondérale, des infections associées et des lésions cutanéo-muqueuses périanales (figure 2).
Figure 2. Lésions anales bourgeonnantes au cours d’une entérocolite par mutation du récepteur à l’IL10.
Des éléments cutanés associés peuvent être présents, notamment des lésions éczématiformes, voire une érythrodermie. Une colite hémorragique associée à une thrombopénie est très évocatrice de syndrome de Wiskott-Aldrich. L’éviction des protéines de lait de vache peut apporter une amélioration partielle et/ou transitoire mais moins franche que dans le cas d’une APLV. Suspecter un déficit immunitaire constitutionnel en cas de :
– allergie alimentaire et signes extradigestifs ;
– entéropathie précoce sévère non-résolutive après éviction des PLV. Entéropathie précoce : penser aux déficits immunitaires !
Présentation pseudo-DIC d'une allergie alimentaire : le syndrome d'Heiner
Le syndrome d’Heiner (milk induced pulmonary syndrome) est une maladie respiratoire chronique associée à la présence d’IgG précipitant en présence de protéines de lait de vache, et résolutive après éviction de l’allergène. Elle s’exprime par des symptômes respiratoires chroniques dans la petite enfance, pouvant en imposer pour des infections récurrentes. Il peut s’agir d’infiltrats pulmonaires en verre dépoli, voire d’hémosidérose pulmonaire responsable d’une anémie microcytaire. L’altération de la croissance peut faire évoquer à tort un DIC, dont le diagnostic est récusé par la normalité des explorations immunologiques et la réversibilité rapide et complète des symptômes après éviction du lait de vache.
Conclusion
Un déficit immunitaire constitutionnel doit être évoqué en cas d’allergie alimentaire associée à des signes extra-digestifs, notamment infectieux ou hématologiques, ou en cas d’entéropathie précoce sévère évocatrice d’allergie aux protéines du lait de vache. Le retard diagnostique peut être préjudiciable : au moindre doute un avis immuno-pédiatrique est indispensable.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :





