Publié le 20 oct 2022Lecture 7 min
Pas qu’une simple gastroentérite...
Chaymae MOUHOUB, Elsa AMOUYAL, Solesne PAPILLARD, Pédiatrie générale et Urgences, HDJ Onco-hématologie, CHU Ambroise-Paré (AP-HP), Boulogne-Billancourt

La rubrique « En direct des staffs » est ouverte à tout médecin d’un service de pédiatrie souhaitant partager avec les lecteurs de Pédiatrie Pratique les cas discutés dans son service et qu’il estime suffisamment intéressants et édifiants pour être portés à la connaissance de ses confrères.
Un garçon de 5 ans est conduit aux urgences pour des douleurs abdominales accompagnées de vomissements alimentaires et de selles liquides non glairo-sanglantes évoluant depuis 48 heures. À l’anamnèse, les parents rapportent une diminution de la prise alimentaire, ainsi qu’une impression de décoloration des selles. Il n’y a pas eu de fièvre, de contage infectieux intrafamilial, ni de voyage récent. Il n’a pas d’antécédent médico-chirurgical et ses vaccinations sont à jour.
Ses constantes à son arrivée aux urgences sont les suivantes : Glasgow :15/15 ; T°: 37,2 °C ; FC : 117/min ; TA : 110/68 mmHg ; SpO2 98 % en air ambiant ; glycémie capillaire : 0,91g/L ; EVA : 6/10.
À l’examen clinique, l’enfant est asthénique, l’hémodynamique périphérique est conservée. L’examen cardiopulmonaire est normal. On note un ictère conjonctival. L’abdomen est distendu et sensible dans son ensemble, sans défense ni contracture avec un débord hépatique à deux travers de doigt. L’examen neurologique est sans particularité.
Le bilan biologique met en évidence une perturbation du bilan hépatique, avec une cytolyse à 6 fois la normale (ASAT : 201 UI/L ; ALAT : 297 UI/L), une cholestase (GGT : 477 UI/L ; PAL : 537 UI/L) et un ictère à bilirubine conjuguée (bilirubine totale 100 μmol/L ; bilirubine conjuguée 89 μmol/L). Il y a également une lipasémie augmentée à 3 837 UI/L, soit 60 fois la normale, et des LDH élevées à 677 UI/L (norme comprise entre 190 et 400 UI/L). L’ionogramme sanguin révèle une insuffisance rénale, avec une urée à 9 mmol/L et une créatinine à 73 μmol/L. Sur le plan inflammatoire, la CRP est à 16 mg/L ; il n’y a pas d’hyperleucocytose. L’échographie abdominale montre un aspect compatible avec une pancréatite aiguë, une hépato-splénomégalie hétérogène, ainsi qu’un épaississement focal hyper-vascularisé d’une anse grêle en para-ombilical.
La prise en charge thérapeutique immédiate a comporté des remplissages vasculaires répétés à l’aide de solutés cristalloïdes, une hyperhydratation intraveineuse, une prise en charge antalgique puis un transfert dans une unité de surveillance continue. En effet, selon les dernières recommandations pédiatriques de 2018 sur les pancréatites aiguës, la prise en charge hémodynamique précoce par des remplissages vasculaires suivie d’une hyperhydratation durant les 24 premières heures, est capitale afin de préserver une perfusion pancréatique suffisante qui prévient la formation de micro-thrombus, et ainsi la survenue de complications locales et systémiques.
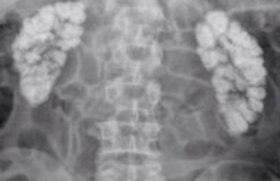
Un complément d’imagerie a été réalisé le lendemain par une IRM abdomino-pelvienne (TDM injectée contre-indiquée en raison de l’insuffisance rénale) qui a montré une importante infiltration tissulaire diffuse intra-et rétropéritonéale évoquant en premier lieu un lymphome (figure).
Figure. IRM abdominale, coupe axiale en séquence T2. Importante infiltration tissulaire intra-et rétropéritonéale.
Des biopsies profondes sous anesthésie générale ont confirmé le diagnostic de lymphome de Burkitt leucémisé. Le bilan d’extension n’a pas montré d’atteinte neuro-méningée (ponction lombaire négative) mais un envahissement médullaire de 30 % au myélogramme. L’hyperhydratation intraveineuse a été poursuivie avec un traitement par rasburicase en prévention du syndrome de lyse tumorale. L’enfant a ensuite été transféré dans un centre de référence en oncologie pédiatrique afin de débuter une chimiothérapie intensive en urgence.
Discussion
Les lymphomes malins sont des proliférations monoclonales malignes de cellules lymphoïdes de degré de maturation variable, à point de départ périphérique, c’est-à-dire non médullaire, contrairement aux leucémies. L’ensemble du système lymphoïde peut être atteint d’emblée ou secondairement (rate, ganglions, amygdales, tube digestif, etc.). Les localisations préférentielles chez l’enfant sont digestives et ORL(1). Selon l’Institut National du cancer (INCA), les lymphomes constituent le 3e cancer le plus fréquent de l’enfant en France avec 195 cas par an, après les leucémies (500 cas par an) et les tumeurs cérébrales (410 cas par an) ; ils représentent 10 % des cancers pédiatriques.
Le lymphome de Burkitt est un lymphome malin non hodgkinien. Il s’agit du lymphome le plus fréquent chez le jeune enfant. Le sex ratio est de 3/1 en faveur du genre masculin, et l’âge de survenue se situe entre 5 et 15 ans. Ce lymphome résulte d’une translocation spécifique t (8 ;14), qui juxtapose l’oncogène c-myc près du gène des chaînes lourdes des immunoglobulines, avec pour conséquence l’activation de l’oncogène et la stimulation du cycle cellulaire. Ainsi, il s’agit le plus souvent de tumeurs avec un indice de prolifération élevé(2).
Les manifestations cliniques sont très variables allant d’une masse abdominale à une constipation ou d’emblée une complication digestive (invagination, occlusion). Il peut aussi se manifester par des adénopathies cervicales unilatérales, une tuméfaction maxillaire, ou une atteinte neuro-méningée d’emblée (méningite, atteinte des paires crâniennes). Plus rarement, comme dans ce cas clinique, le lymphome peut se révéler par un tableau de pancréatite aiguë en cas d’infiltration diffuse du pancréas avec un épaississement des voies biliaires et comme manifestations clinico-biologiques des douleurs abdominales, des vomissements, des selles liquides, un ictère avec perturbation du bilan hépatique(1,3).
Le diagnostic est histologique et les prélèvements doivent être le moins invasifs possibles en premier lieu (ponction d’épanchement, myélogramme, biopsie ganglionnaire, voire biopsies profondes en milieu oncologique spécialisé).
Le bilan initial comprend tout d’abord un bilan biologique afin d’évaluer l’intensité du syndrome de lyse tumorale : lactate deshydrogénase (LDH), ionogramme sanguin, fonction rénale, phosphorémie, calcémie et uricémie. Le dosage du taux de LDH au diagnostic est capital et présente plusieurs intérêts ; un taux élevé de LDH témoigne d’un indice de prolifération élevé de la tumeur, ce qui représente un facteur de risque de lyse tumorale. Un taux élevé au diagnostic a également été associé à un facteur de mauvais pronostic sur la réponse au traitement. De plus, il présente un intérêt pour le suivi de la maladie. Il est important de noter que la LDH est une enzyme cytoplasmique présente dans tous les tissus et est facilement libérée au niveau plasmatique en cas de lésion tissulaire de façon non spécifique. En effet, une augmentation de la LDH peut être retrouvée en cas d’hépatite, d’anémie hémolytique, de pathologie rénale, de maladies musculaires, etc.
Le bilan d’extension comporte : un scanner thoraco-abdominal à la recherche d’une atteinte médiastinale et hépato-splénique, un myélogramme à la recherche d’un envahissement de la moelle osseuse, une biopsie ostéo-médullaire et une ponction lombaire à la recherche d’une atteinte méningée.
Les lymphomes de Burkitt sont classés en quatre stades selon l’étendue de leur localisation, l’accessibilité à une résection chirurgicale éventuelle et la présence d’un envahissement médullaire ou neuro-méningé. Cette classification permet de déterminer le pronostic et la prise en charge selon trois groupes de traitement(4).
Le traitement doit être débuté en urgence par une polychimiothérapie intensive. Une chirurgie peut être réalisée en cas de tumeur localisée de petite taille après accord d’un centre de référence. Une association avec un anticorps monoclonal (rituximab) est indiquée dans certains groupes plus sévères de patients. Le rituximab se lie spécifiquement à l’antigène transmembranaire CD20, une phosphoprotéine située sur les lymphocytes pré-B et B matures qui s’exprime dans plus de 95 % des cellules B des lymphomes non hodgkiniens. Le traitement est court mais intensif, et adapté au stade de la maladie(5).
Ce sont des tumeurs de très bon pronostic en raison de leur très grande chimiosensibilité et de l’association du rituximab aux chimiothérapies pour les formes les plus sévères. La survie sans récidive à 5 ans se situe entre 80 à 100 % selon le stade. Les récidives surviennent le plus souvent la première année avec dans ce cas, un taux de survie aux alentours de 30 %.
Points clés
Il faut savoir évoquer un lymphome de Burkitt abdominal en présence d’une masse abdominale, d’une invagination intestinale aiguë en dehors des âges typiques ou en cas d’association à un syndrome tumoral, ainsi qu’à l’imagerie devant la présence d’ascite, d’anses digestives épaissies ou infiltrées.
Les lymphomes de Burkitt constituent une urgence diagnostique et thérapeutique car ce sont des tumeurs agressives mais de bon pronostic. Il faut par ailleurs prévenir le syndrome de lyse tumorale qui est souvent important et peut mettre en jeu initialement le pronostic vital.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :